Request Demo
What are BCL11A gene inhibitors and how do they work?
21 June 2024
The BCL11A gene has emerged as a significant player in the field of genetic research, especially in relation to hemoglobinopathies such as sickle cell disease and beta-thalassemia. The discovery and subsequent development of BCL11A gene inhibitors have revolutionized the approach to treating these inherited blood disorders. These inhibitors offer a promising avenue for therapeutic intervention by targeting the genetic pathways that regulate the production of different types of hemoglobin. In this blog post, we will delve into the role of BCL11A gene inhibitors, their mechanisms of action, and their clinical applications.
BCL11A, or B-cell CLL/lymphoma 11A, is a transcription factor that plays a crucial role in the regulation of globin gene expression. Globin genes are responsible for the production of hemoglobin, the protein in red blood cells that transports oxygen throughout the body. There are different types of hemoglobin produced during various stages of human development: embryonic, fetal, and adult hemoglobin. BCL11A is primarily involved in the switch from fetal hemoglobin (HbF) to adult hemoglobin (HbA) production after birth. Elevated levels of HbF have been shown to ameliorate the clinical severity of hemoglobinopathies. Thus, inhibiting BCL11A has the potential to sustain HbF production in adults, providing a therapeutic strategy for conditions characterized by defective or deficient adult hemoglobin.
BCL11A gene inhibitors work by disrupting the activity of the BCL11A transcription factor. This disruption can be achieved through various mechanisms, including small molecules, antisense oligonucleotides, and CRISPR/Cas9 gene editing. Small molecule inhibitors bind to the BCL11A protein, altering its conformation and preventing it from binding to DNA. Antisense oligonucleotides, on the other hand, are short strands of DNA or RNA that bind to the mRNA transcripts of BCL11A, leading to their degradation and reducing the synthesis of the BCL11A protein. CRISPR/Cas9 gene editing technology can be employed to directly target and disrupt the BCL11A gene within the genome, thereby permanently reducing its expression.
The inhibition of BCL11A results in the reactivation of fetal hemoglobin production in adults. This reactivation occurs because BCL11A normally acts as a repressor of the gamma-globin genes, which are responsible for HbF production. By inhibiting BCL11A, the repression is lifted, allowing for the increased expression of gamma-globin and the subsequent production of HbF. This increased level of HbF can compensate for the defective hemoglobin present in individuals with sickle cell disease or beta-thalassemia, alleviating the symptoms and improving their quality of life.
BCL11A gene inhibitors have shown great promise in the treatment of hemoglobinopathies such as sickle cell disease and beta-thalassemia. Sickle cell disease is characterized by the production of abnormal hemoglobin, which causes red blood cells to become rigid and sickle-shaped, leading to painful episodes, anemia, and organ damage. Beta-thalassemia, on the other hand, is caused by mutations in the beta-globin gene, resulting in reduced or absent production of beta-globin chains, leading to ineffective erythropoiesis and severe anemia. By reactivating HbF production, BCL11A inhibitors can mitigate the effects of these disorders.
Several clinical trials are currently underway to evaluate the safety and efficacy of BCL11A gene inhibitors. Early results from these trials have been promising, with patients showing increased levels of HbF and improvements in clinical symptoms. Moreover, the use of gene editing technologies such as CRISPR/Cas9 has demonstrated the potential for long-term and possibly curative treatments by permanently modifying the patient's genetic makeup to produce HbF.
In conclusion, BCL11A gene inhibitors represent a groundbreaking advancement in the treatment of hemoglobinopathies. By targeting the genetic pathways that regulate hemoglobin production, these inhibitors offer a novel therapeutic strategy that could significantly improve the lives of individuals suffering from sickle cell disease and beta-thalassemia. As research and clinical trials continue to progress, there is hope that BCL11A gene inhibitors will become a cornerstone of treatment for these debilitating conditions.
BCL11A, or B-cell CLL/lymphoma 11A, is a transcription factor that plays a crucial role in the regulation of globin gene expression. Globin genes are responsible for the production of hemoglobin, the protein in red blood cells that transports oxygen throughout the body. There are different types of hemoglobin produced during various stages of human development: embryonic, fetal, and adult hemoglobin. BCL11A is primarily involved in the switch from fetal hemoglobin (HbF) to adult hemoglobin (HbA) production after birth. Elevated levels of HbF have been shown to ameliorate the clinical severity of hemoglobinopathies. Thus, inhibiting BCL11A has the potential to sustain HbF production in adults, providing a therapeutic strategy for conditions characterized by defective or deficient adult hemoglobin.
BCL11A gene inhibitors work by disrupting the activity of the BCL11A transcription factor. This disruption can be achieved through various mechanisms, including small molecules, antisense oligonucleotides, and CRISPR/Cas9 gene editing. Small molecule inhibitors bind to the BCL11A protein, altering its conformation and preventing it from binding to DNA. Antisense oligonucleotides, on the other hand, are short strands of DNA or RNA that bind to the mRNA transcripts of BCL11A, leading to their degradation and reducing the synthesis of the BCL11A protein. CRISPR/Cas9 gene editing technology can be employed to directly target and disrupt the BCL11A gene within the genome, thereby permanently reducing its expression.
The inhibition of BCL11A results in the reactivation of fetal hemoglobin production in adults. This reactivation occurs because BCL11A normally acts as a repressor of the gamma-globin genes, which are responsible for HbF production. By inhibiting BCL11A, the repression is lifted, allowing for the increased expression of gamma-globin and the subsequent production of HbF. This increased level of HbF can compensate for the defective hemoglobin present in individuals with sickle cell disease or beta-thalassemia, alleviating the symptoms and improving their quality of life.
BCL11A gene inhibitors have shown great promise in the treatment of hemoglobinopathies such as sickle cell disease and beta-thalassemia. Sickle cell disease is characterized by the production of abnormal hemoglobin, which causes red blood cells to become rigid and sickle-shaped, leading to painful episodes, anemia, and organ damage. Beta-thalassemia, on the other hand, is caused by mutations in the beta-globin gene, resulting in reduced or absent production of beta-globin chains, leading to ineffective erythropoiesis and severe anemia. By reactivating HbF production, BCL11A inhibitors can mitigate the effects of these disorders.
Several clinical trials are currently underway to evaluate the safety and efficacy of BCL11A gene inhibitors. Early results from these trials have been promising, with patients showing increased levels of HbF and improvements in clinical symptoms. Moreover, the use of gene editing technologies such as CRISPR/Cas9 has demonstrated the potential for long-term and possibly curative treatments by permanently modifying the patient's genetic makeup to produce HbF.
In conclusion, BCL11A gene inhibitors represent a groundbreaking advancement in the treatment of hemoglobinopathies. By targeting the genetic pathways that regulate hemoglobin production, these inhibitors offer a novel therapeutic strategy that could significantly improve the lives of individuals suffering from sickle cell disease and beta-thalassemia. As research and clinical trials continue to progress, there is hope that BCL11A gene inhibitors will become a cornerstone of treatment for these debilitating conditions.
How to obtain the latest development progress of all targets?
In the Synapse database, you can stay updated on the latest research and development advances of all targets. This service is accessible anytime and anywhere, with updates available daily or weekly. Use the "Set Alert" function to stay informed. Click on the image below to embark on a brand new journey of drug discovery!
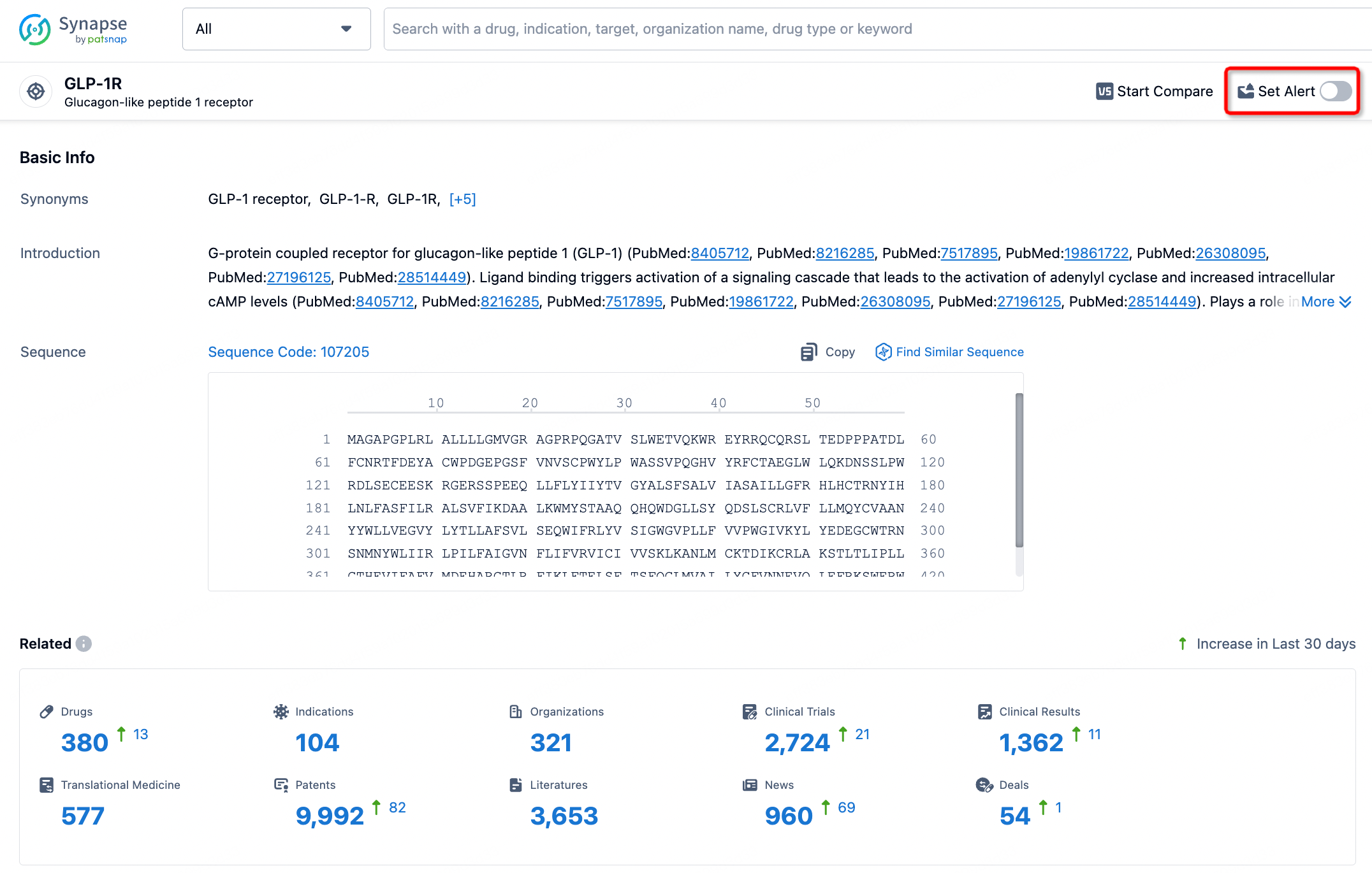
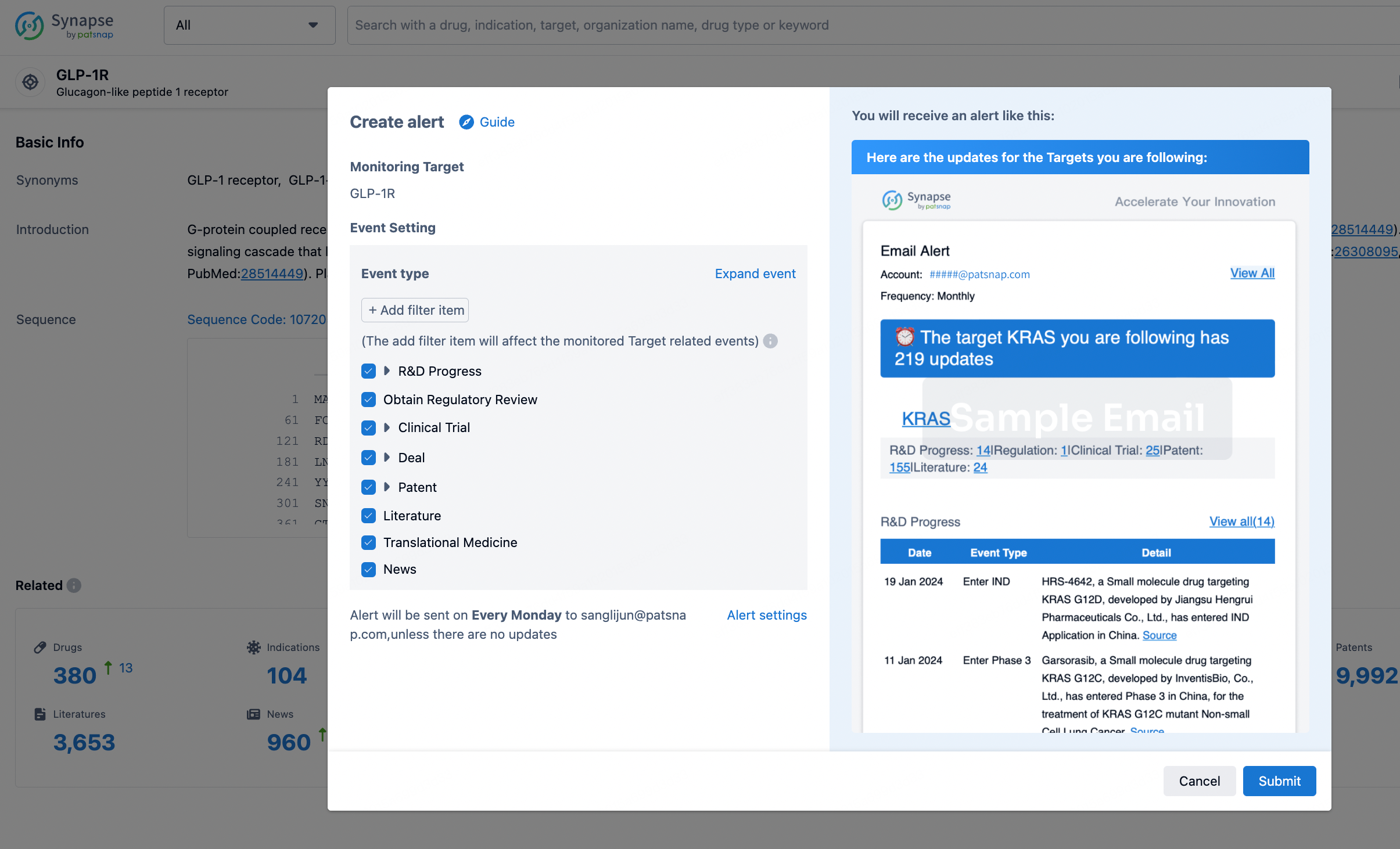
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.