Request Demo
What are CAG repeat expansion inhibitors and how do they work?
25 June 2024
CAG repeat expansion disorders, such as Huntington’s disease, spinocerebellar ataxias, and certain types of myotonic dystrophy, are genetic conditions characterized by the abnormal repetition of the CAG trinucleotide sequence in specific genes. These repeated sequences cause the production of faulty proteins that lead to cellular dysfunction and, ultimately, disease symptoms. In recent years, researchers have been focusing on developing therapeutic strategies to combat these disorders, one of which is the use of CAG repeat expansion inhibitors.
CAG repeat expansion inhibitors are a class of compounds designed to target the root cause of these genetic conditions: the expanded CAG repeats in the DNA. By interfering with the expression of these expanded repeats, these inhibitors aim to reduce or halt the production of toxic proteins that lead to disease progression. Understanding how these inhibitors work provides valuable insight into their potential to revolutionize the treatment of CAG repeat expansion disorders.
CAG repeat expansion inhibitors work by various mechanisms aimed at reducing the toxic effects of the mutant proteins produced by expanded CAG repeats. One common approach involves the use of antisense oligonucleotides (ASOs). These are short, synthetic strands of nucleic acids that are designed to bind specifically to the mutant mRNA transcripts produced by the expanded CAG repeats. By binding to these transcripts, ASOs can promote their degradation or prevent their translation into faulty proteins. This reduces the overall level of toxic proteins in the cells, potentially alleviating disease symptoms.
Another approach involves small molecules that can specifically bind to the expanded CAG repeats in the DNA or RNA. These small molecules can disrupt the formation of abnormal secondary structures that are characteristic of expanded CAG repeats, such as hairpins or R-loops. By preventing these structures from forming, small molecules can reduce the stability and persistence of the mutant transcripts and proteins, thereby mitigating their toxic effects.
Gene editing technologies, such as CRISPR/Cas9, also hold promise as a tool for inhibiting CAG repeat expansions. By selectively targeting and editing the expanded CAG repeats in the DNA, these technologies can potentially correct the underlying genetic mutation. While still in the early stages of development, gene editing represents a powerful strategy for addressing the root cause of CAG repeat expansion disorders.
CAG repeat expansion inhibitors are primarily used in the context of neurodegenerative and neuromuscular disorders, where expanded CAG repeats play a central role in disease pathology. Huntington’s disease (HD) is one of the most well-known conditions associated with CAG repeat expansions. HD is a progressive neurodegenerative disorder characterized by motor dysfunction, cognitive decline, and psychiatric symptoms. There is currently no cure for HD, and available treatments only address symptoms without modifying the disease course. CAG repeat expansion inhibitors hold significant promise as a disease-modifying therapy for HD by targeting the production of the toxic huntingtin protein.
Spinocerebellar ataxias (SCAs) are another group of disorders associated with expanded CAG repeats. SCAs are characterized by progressive degeneration of the cerebellum and other parts of the nervous system, leading to impaired coordination and balance. Similar to HD, there are currently no disease-modifying treatments for SCAs. CAG repeat expansion inhibitors could potentially slow or halt the progression of these disorders by reducing the levels of toxic proteins associated with the expanded repeats.
Certain forms of myotonic dystrophy, particularly myotonic dystrophy type 1 (DM1), are also caused by expanded CAG repeats. DM1 is a multisystem disorder that affects skeletal and cardiac muscles, as well as other tissues. Current treatments for DM1 are largely supportive and do not address the underlying genetic cause. CAG repeat expansion inhibitors have the potential to provide a targeted therapeutic approach for DM1 by reducing the production of toxic RNA transcripts and proteins.
In conclusion, CAG repeat expansion inhibitors represent a promising avenue for the treatment of genetic disorders caused by expanded CAG repeats. Through various mechanisms, these inhibitors aim to reduce the production and toxic effects of mutant proteins, offering hope for disease-modifying therapies. As research in this field progresses, CAG repeat expansion inhibitors may become a crucial component of the therapeutic arsenal for conditions such as Huntington’s disease, spinocerebellar ataxias, and myotonic dystrophy.
CAG repeat expansion inhibitors are a class of compounds designed to target the root cause of these genetic conditions: the expanded CAG repeats in the DNA. By interfering with the expression of these expanded repeats, these inhibitors aim to reduce or halt the production of toxic proteins that lead to disease progression. Understanding how these inhibitors work provides valuable insight into their potential to revolutionize the treatment of CAG repeat expansion disorders.
CAG repeat expansion inhibitors work by various mechanisms aimed at reducing the toxic effects of the mutant proteins produced by expanded CAG repeats. One common approach involves the use of antisense oligonucleotides (ASOs). These are short, synthetic strands of nucleic acids that are designed to bind specifically to the mutant mRNA transcripts produced by the expanded CAG repeats. By binding to these transcripts, ASOs can promote their degradation or prevent their translation into faulty proteins. This reduces the overall level of toxic proteins in the cells, potentially alleviating disease symptoms.
Another approach involves small molecules that can specifically bind to the expanded CAG repeats in the DNA or RNA. These small molecules can disrupt the formation of abnormal secondary structures that are characteristic of expanded CAG repeats, such as hairpins or R-loops. By preventing these structures from forming, small molecules can reduce the stability and persistence of the mutant transcripts and proteins, thereby mitigating their toxic effects.
Gene editing technologies, such as CRISPR/Cas9, also hold promise as a tool for inhibiting CAG repeat expansions. By selectively targeting and editing the expanded CAG repeats in the DNA, these technologies can potentially correct the underlying genetic mutation. While still in the early stages of development, gene editing represents a powerful strategy for addressing the root cause of CAG repeat expansion disorders.
CAG repeat expansion inhibitors are primarily used in the context of neurodegenerative and neuromuscular disorders, where expanded CAG repeats play a central role in disease pathology. Huntington’s disease (HD) is one of the most well-known conditions associated with CAG repeat expansions. HD is a progressive neurodegenerative disorder characterized by motor dysfunction, cognitive decline, and psychiatric symptoms. There is currently no cure for HD, and available treatments only address symptoms without modifying the disease course. CAG repeat expansion inhibitors hold significant promise as a disease-modifying therapy for HD by targeting the production of the toxic huntingtin protein.
Spinocerebellar ataxias (SCAs) are another group of disorders associated with expanded CAG repeats. SCAs are characterized by progressive degeneration of the cerebellum and other parts of the nervous system, leading to impaired coordination and balance. Similar to HD, there are currently no disease-modifying treatments for SCAs. CAG repeat expansion inhibitors could potentially slow or halt the progression of these disorders by reducing the levels of toxic proteins associated with the expanded repeats.
Certain forms of myotonic dystrophy, particularly myotonic dystrophy type 1 (DM1), are also caused by expanded CAG repeats. DM1 is a multisystem disorder that affects skeletal and cardiac muscles, as well as other tissues. Current treatments for DM1 are largely supportive and do not address the underlying genetic cause. CAG repeat expansion inhibitors have the potential to provide a targeted therapeutic approach for DM1 by reducing the production of toxic RNA transcripts and proteins.
In conclusion, CAG repeat expansion inhibitors represent a promising avenue for the treatment of genetic disorders caused by expanded CAG repeats. Through various mechanisms, these inhibitors aim to reduce the production and toxic effects of mutant proteins, offering hope for disease-modifying therapies. As research in this field progresses, CAG repeat expansion inhibitors may become a crucial component of the therapeutic arsenal for conditions such as Huntington’s disease, spinocerebellar ataxias, and myotonic dystrophy.
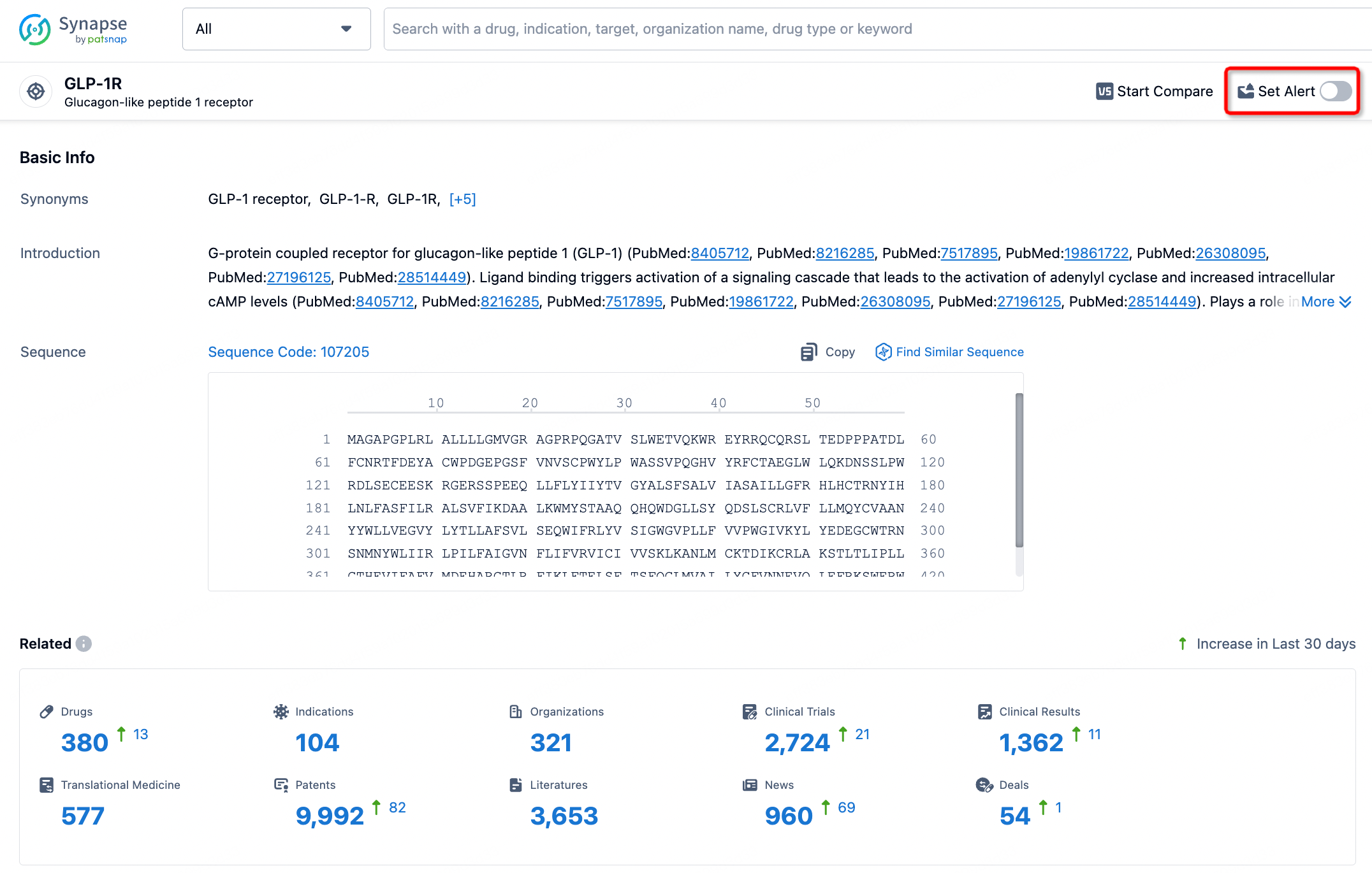
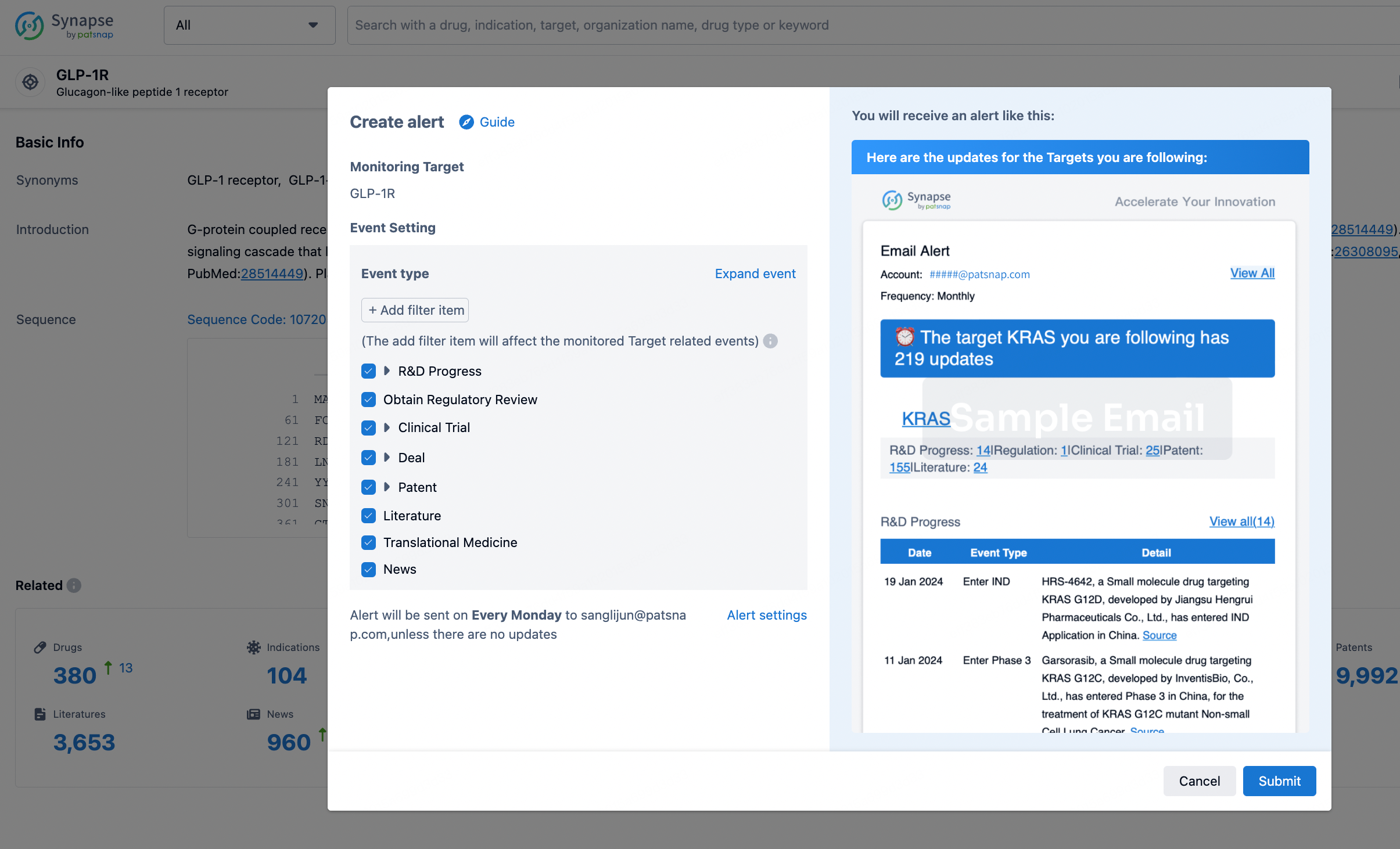
How to obtain the latest development progress of all targets?
In the Synapse database, you can stay updated on the latest research and development advances of all targets. This service is accessible anytime and anywhere, with updates available daily or weekly. Use the "Set Alert" function to stay informed. Click on the image below to embark on a brand new journey of drug discovery!
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.