Request Demo
What are Huntingtin inhibitors and how do they work?
21 June 2024
Huntington’s disease (HD) is a devastating neurodegenerative disorder caused by a mutation in the huntingtin gene (HTT). This mutation results in the production of an abnormally long and toxic version of the huntingtin protein, which leads to the progressive degeneration of neurons in the brain. As scientists and researchers continue to search for effective treatments for HD, one promising avenue is the development of Huntingtin inhibitors. These compounds aim to reduce the levels of the harmful huntingtin protein, offering hope for slowing down or potentially halting the progression of the disease.
Huntingtin inhibitors are a class of therapeutic agents designed to interfere with the production, aggregation, or function of the mutant huntingtin protein. The idea is to reduce the toxic effects of the protein and thereby mitigate the neuronal damage it causes. Various approaches to inhibiting huntingtin have been explored, including small molecules, RNA-based therapies, and gene editing technologies.
At the core of many Huntingtin inhibitors is the concept of gene silencing, where the expression of the mutant HTT gene is reduced. RNA interference (RNAi) and antisense oligonucleotides (ASOs) are two prominent techniques employed in this strategy. RNAi uses small interfering RNAs (siRNAs) to target and degrade the messenger RNA (mRNA) that codes for the mutant huntingtin protein. By breaking down this mRNA, RNAi effectively reduces the protein’s production.
On the other hand, ASOs are short, synthetic strands of DNA designed to bind to the mRNA of the mutant huntingtin gene. This binding blocks the translation of the mRNA into the protein, thereby decreasing the levels of the toxic huntingtin protein. These approaches work at the genetic level, providing a powerful means to tackle the root cause of HD.
Another intriguing method involves the use of small molecules to inhibit the aggregation of the mutant huntingtin protein. Aggregation is a process where the protein misfolds and forms clumps, which are toxic to neurons. Small molecules can interfere with this aggregation, helping to maintain protein homeostasis and protect neuronal health.
Huntingtin inhibitors are primarily being developed to treat Huntington’s disease. HD is a rare, inherited disorder that usually manifests in mid-adulthood, leading to a gradual decline in motor control, cognitive function, and mental health. The progressive nature of the disease means that symptoms worsen over time, ultimately resulting in severe disability and early death. There is currently no cure for HD, and available treatments only address symptoms rather than halting disease progression.
The goal of Huntingtin inhibitors is to reduce the levels of the mutant huntingtin protein and thereby slow or stop the progression of HD. By targeting the production or aggregation of the toxic protein, these inhibitors aim to preserve neuronal function and delay the onset of symptoms. Early clinical trials have shown promise, with some RNAi and ASO-based therapies demonstrating the ability to lower huntingtin levels in the brain and improve motor and cognitive functions in preclinical models.
In addition to HD, there is potential for Huntingtin inhibitors to be applied to other neurodegenerative disorders that share similar pathological mechanisms. For instance, certain forms of spinocerebellar ataxia (SCA) and amyotrophic lateral sclerosis (ALS) involve toxic protein aggregation, and strategies developed for HD may be adapted to target these diseases. Furthermore, the advancements in gene silencing and protein aggregation inhibition techniques could pave the way for novel treatments for a range of neurological conditions.
In conclusion, Huntingtin inhibitors represent a promising frontier in the fight against Huntington’s disease. By targeting the root cause of the disorder—the mutant huntingtin protein—these therapies hold the potential to significantly alter the disease’s course. While challenges remain in terms of delivery, specificity, and long-term effects, ongoing research and clinical trials are continually advancing our understanding and bringing us closer to effective treatments. The hope is that, one day, Huntington’s disease and other similar neurodegenerative disorders can be effectively managed, offering patients and families a brighter future.
Huntingtin inhibitors are a class of therapeutic agents designed to interfere with the production, aggregation, or function of the mutant huntingtin protein. The idea is to reduce the toxic effects of the protein and thereby mitigate the neuronal damage it causes. Various approaches to inhibiting huntingtin have been explored, including small molecules, RNA-based therapies, and gene editing technologies.
At the core of many Huntingtin inhibitors is the concept of gene silencing, where the expression of the mutant HTT gene is reduced. RNA interference (RNAi) and antisense oligonucleotides (ASOs) are two prominent techniques employed in this strategy. RNAi uses small interfering RNAs (siRNAs) to target and degrade the messenger RNA (mRNA) that codes for the mutant huntingtin protein. By breaking down this mRNA, RNAi effectively reduces the protein’s production.
On the other hand, ASOs are short, synthetic strands of DNA designed to bind to the mRNA of the mutant huntingtin gene. This binding blocks the translation of the mRNA into the protein, thereby decreasing the levels of the toxic huntingtin protein. These approaches work at the genetic level, providing a powerful means to tackle the root cause of HD.
Another intriguing method involves the use of small molecules to inhibit the aggregation of the mutant huntingtin protein. Aggregation is a process where the protein misfolds and forms clumps, which are toxic to neurons. Small molecules can interfere with this aggregation, helping to maintain protein homeostasis and protect neuronal health.
Huntingtin inhibitors are primarily being developed to treat Huntington’s disease. HD is a rare, inherited disorder that usually manifests in mid-adulthood, leading to a gradual decline in motor control, cognitive function, and mental health. The progressive nature of the disease means that symptoms worsen over time, ultimately resulting in severe disability and early death. There is currently no cure for HD, and available treatments only address symptoms rather than halting disease progression.
The goal of Huntingtin inhibitors is to reduce the levels of the mutant huntingtin protein and thereby slow or stop the progression of HD. By targeting the production or aggregation of the toxic protein, these inhibitors aim to preserve neuronal function and delay the onset of symptoms. Early clinical trials have shown promise, with some RNAi and ASO-based therapies demonstrating the ability to lower huntingtin levels in the brain and improve motor and cognitive functions in preclinical models.
In addition to HD, there is potential for Huntingtin inhibitors to be applied to other neurodegenerative disorders that share similar pathological mechanisms. For instance, certain forms of spinocerebellar ataxia (SCA) and amyotrophic lateral sclerosis (ALS) involve toxic protein aggregation, and strategies developed for HD may be adapted to target these diseases. Furthermore, the advancements in gene silencing and protein aggregation inhibition techniques could pave the way for novel treatments for a range of neurological conditions.
In conclusion, Huntingtin inhibitors represent a promising frontier in the fight against Huntington’s disease. By targeting the root cause of the disorder—the mutant huntingtin protein—these therapies hold the potential to significantly alter the disease’s course. While challenges remain in terms of delivery, specificity, and long-term effects, ongoing research and clinical trials are continually advancing our understanding and bringing us closer to effective treatments. The hope is that, one day, Huntington’s disease and other similar neurodegenerative disorders can be effectively managed, offering patients and families a brighter future.
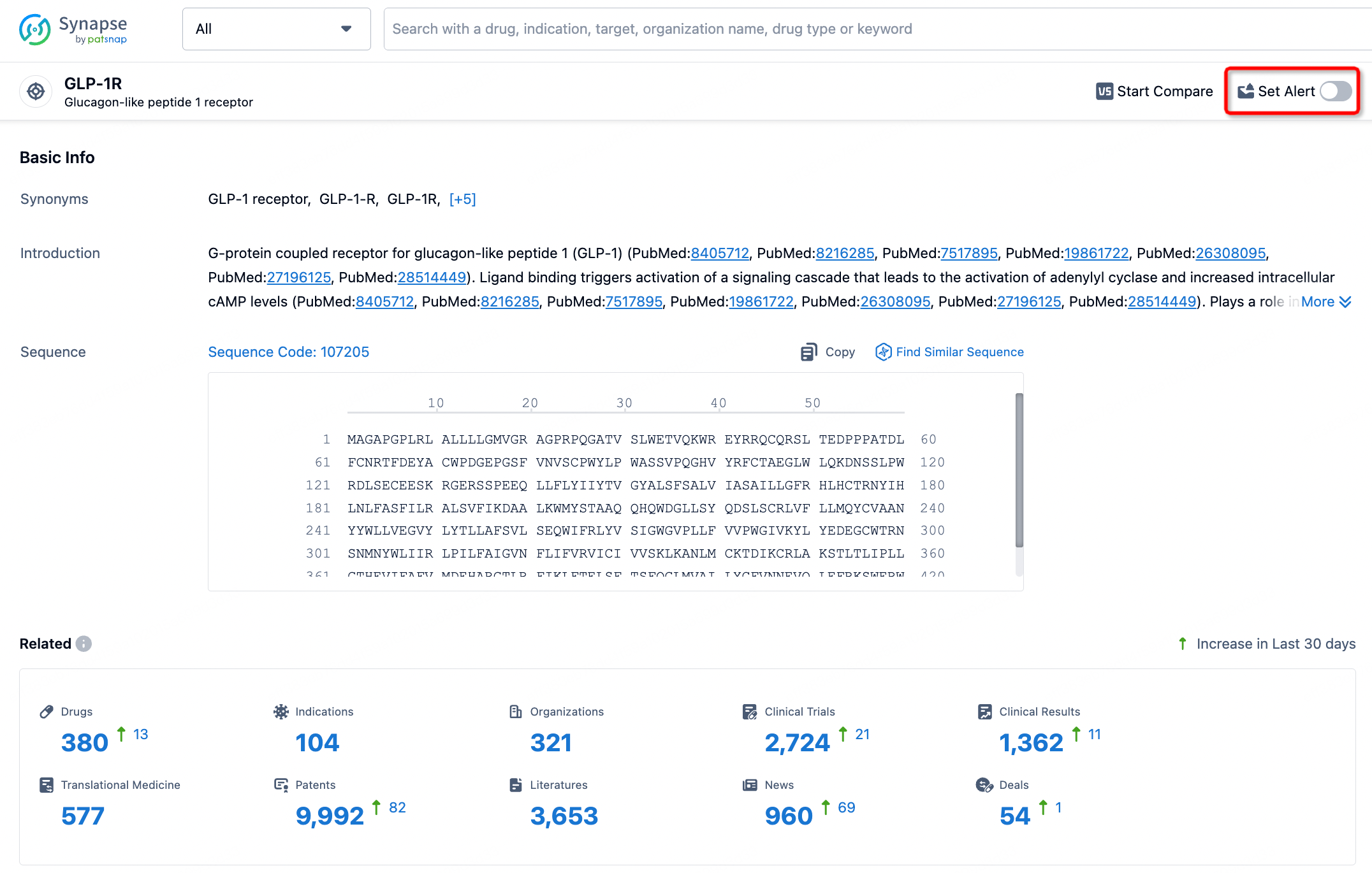
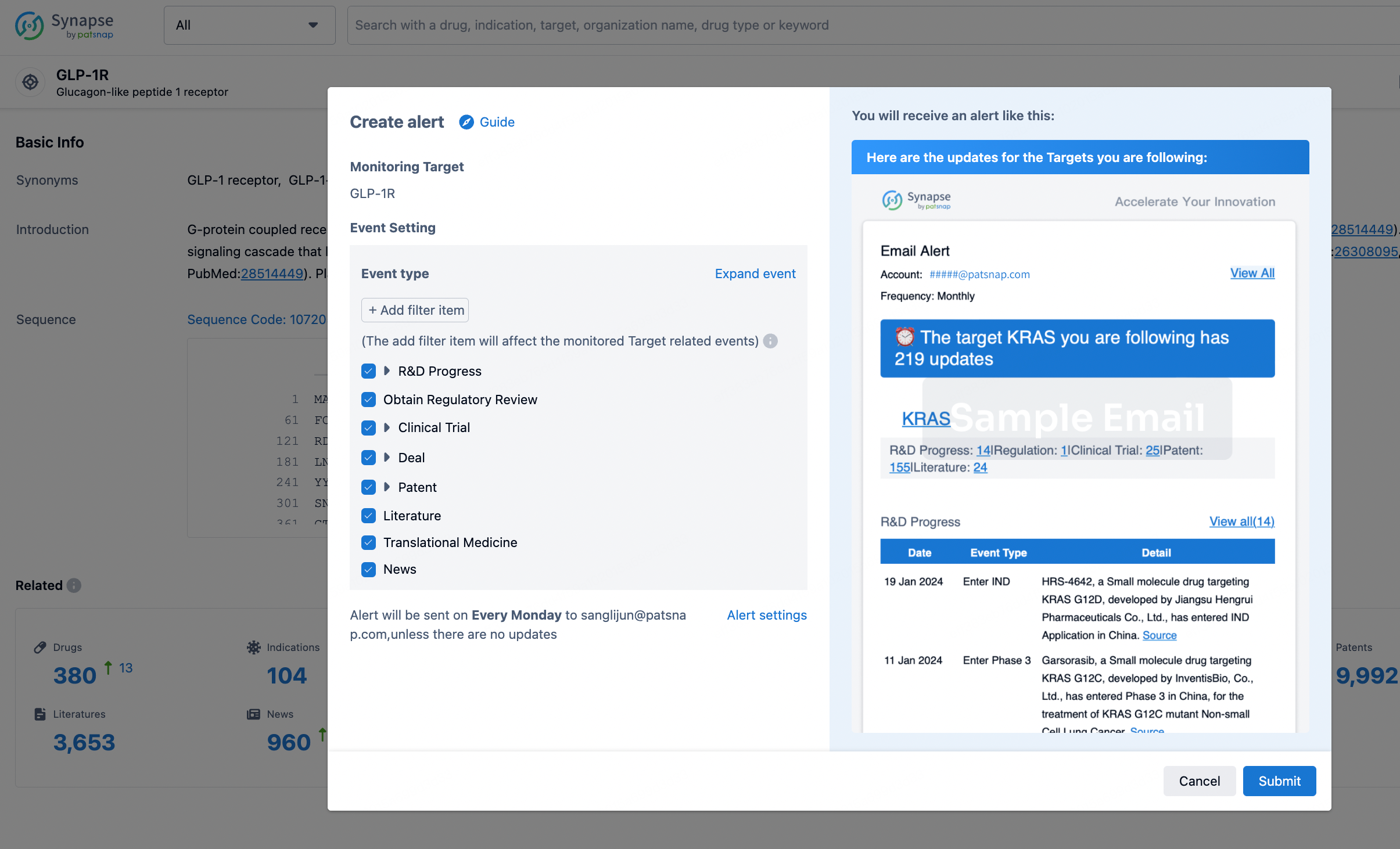
How to obtain the latest development progress of all targets?
In the Synapse database, you can stay updated on the latest research and development advances of all targets. This service is accessible anytime and anywhere, with updates available daily or weekly. Use the "Set Alert" function to stay informed. Click on the image below to embark on a brand new journey of drug discovery!
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.