Request Demo
What are Iduronate sulfatase replacements and how do they work?
21 June 2024
Iduronate sulfatase, also known as IDS, is an essential enzyme involved in the breakdown of glycosaminoglycans (GAGs) such as dermatan sulfate and heparan sulfate within the lysosomes of cells. This process is crucial for maintaining cellular function and preventing the accumulation of harmful substances that can disrupt cellular processes. When there is a deficiency or malfunction in the production of iduronate sulfatase, it results in a group of lysosomal storage disorders, with Hunter syndrome (mucopolysaccharidosis type II) being the most prominent. This genetic disorder leads to severe physical and neurological impairments. Fortunately, advancements in medical science have introduced iduronate sulfatase replacements, offering a lifeline for patients with these conditions.
Iduronate sulfatase replacements are a form of enzyme replacement therapy (ERT) designed to substitute the deficient or malfunctioning enzyme in patients with Hunter syndrome. These therapeutic agents are typically produced using recombinant DNA technology, which involves cloning the human iduronate sulfatase gene and expressing it in a host system, often Chinese hamster ovary (CHO) cells. The resulting recombinant enzyme is then purified and formulated for administration to patients.
Once administered, usually through intravenous infusion, the recombinant iduronate sulfatase is distributed throughout the body via the bloodstream. Its primary target is the lysosomes – cellular organelles responsible for breaking down waste materials and cellular debris. The recombinant enzyme enters these organelles and compensates for the deficient natural enzyme, facilitating the breakdown of GAGs. This process helps to reduce the accumulation of these substances, thereby alleviating the symptoms and slowing the progression of the disease.
The effectiveness of iduronate sulfatase replacements largely depends on the ability of the recombinant enzyme to reach and be taken up by cells throughout the body, particularly those in organs most affected by GAG accumulation, such as the liver, spleen, heart, and brain. The uptake mechanism typically involves mannose-6-phosphate receptors on the cell surface, which recognize and internalize the enzyme for delivery to the lysosomes.
Iduronate sulfatase replacements are primarily used for the treatment of Hunter syndrome, a rare and inherited lysosomal storage disorder caused by a deficiency of iduronate sulfatase. Hunter syndrome predominantly affects males and can present with a wide range of symptoms, including developmental delays, behavioral issues, hearing loss, joint stiffness, skeletal abnormalities, and organomegaly. Without treatment, the disease can lead to severe debilitating conditions and a significantly shortened lifespan.
By providing exogenous iduronate sulfatase, ERT aims to restore the normal catabolic processes within the lysosomes, thereby reducing the buildup of harmful GAGs. This therapeutic approach has been shown to improve various clinical outcomes, including enhanced joint mobility, reduced organ size, and improved respiratory function. Furthermore, ERT can contribute to a better quality of life by alleviating some of the physical and functional impairments associated with the disease.
While iduronate sulfatase replacements represent a significant advancement in the management of Hunter syndrome, they are not without limitations. For example, the blood-brain barrier presents a significant challenge for the delivery of the enzyme to the central nervous system (CNS), which means that neurological symptoms of Hunter syndrome may not be fully addressed by current ERTs. Ongoing research aims to develop more effective delivery methods or alternative treatments that can overcome this barrier and provide comprehensive care for patients with CNS involvement.
In conclusion, iduronate sulfatase replacements have revolutionized the treatment landscape for Hunter syndrome, offering hope and improved outcomes for affected individuals. By compensating for the deficient enzyme and reducing the pathological accumulation of GAGs, ERT can significantly mitigate the disease's impact on patients' lives. However, continued research and innovation are essential to address the remaining challenges and ensure that all aspects of Hunter syndrome can be effectively managed. As our understanding of lysosomal storage disorders advances, so too will our ability to treat and potentially cure these debilitating conditions.
Iduronate sulfatase replacements are a form of enzyme replacement therapy (ERT) designed to substitute the deficient or malfunctioning enzyme in patients with Hunter syndrome. These therapeutic agents are typically produced using recombinant DNA technology, which involves cloning the human iduronate sulfatase gene and expressing it in a host system, often Chinese hamster ovary (CHO) cells. The resulting recombinant enzyme is then purified and formulated for administration to patients.
Once administered, usually through intravenous infusion, the recombinant iduronate sulfatase is distributed throughout the body via the bloodstream. Its primary target is the lysosomes – cellular organelles responsible for breaking down waste materials and cellular debris. The recombinant enzyme enters these organelles and compensates for the deficient natural enzyme, facilitating the breakdown of GAGs. This process helps to reduce the accumulation of these substances, thereby alleviating the symptoms and slowing the progression of the disease.
The effectiveness of iduronate sulfatase replacements largely depends on the ability of the recombinant enzyme to reach and be taken up by cells throughout the body, particularly those in organs most affected by GAG accumulation, such as the liver, spleen, heart, and brain. The uptake mechanism typically involves mannose-6-phosphate receptors on the cell surface, which recognize and internalize the enzyme for delivery to the lysosomes.
Iduronate sulfatase replacements are primarily used for the treatment of Hunter syndrome, a rare and inherited lysosomal storage disorder caused by a deficiency of iduronate sulfatase. Hunter syndrome predominantly affects males and can present with a wide range of symptoms, including developmental delays, behavioral issues, hearing loss, joint stiffness, skeletal abnormalities, and organomegaly. Without treatment, the disease can lead to severe debilitating conditions and a significantly shortened lifespan.
By providing exogenous iduronate sulfatase, ERT aims to restore the normal catabolic processes within the lysosomes, thereby reducing the buildup of harmful GAGs. This therapeutic approach has been shown to improve various clinical outcomes, including enhanced joint mobility, reduced organ size, and improved respiratory function. Furthermore, ERT can contribute to a better quality of life by alleviating some of the physical and functional impairments associated with the disease.
While iduronate sulfatase replacements represent a significant advancement in the management of Hunter syndrome, they are not without limitations. For example, the blood-brain barrier presents a significant challenge for the delivery of the enzyme to the central nervous system (CNS), which means that neurological symptoms of Hunter syndrome may not be fully addressed by current ERTs. Ongoing research aims to develop more effective delivery methods or alternative treatments that can overcome this barrier and provide comprehensive care for patients with CNS involvement.
In conclusion, iduronate sulfatase replacements have revolutionized the treatment landscape for Hunter syndrome, offering hope and improved outcomes for affected individuals. By compensating for the deficient enzyme and reducing the pathological accumulation of GAGs, ERT can significantly mitigate the disease's impact on patients' lives. However, continued research and innovation are essential to address the remaining challenges and ensure that all aspects of Hunter syndrome can be effectively managed. As our understanding of lysosomal storage disorders advances, so too will our ability to treat and potentially cure these debilitating conditions.
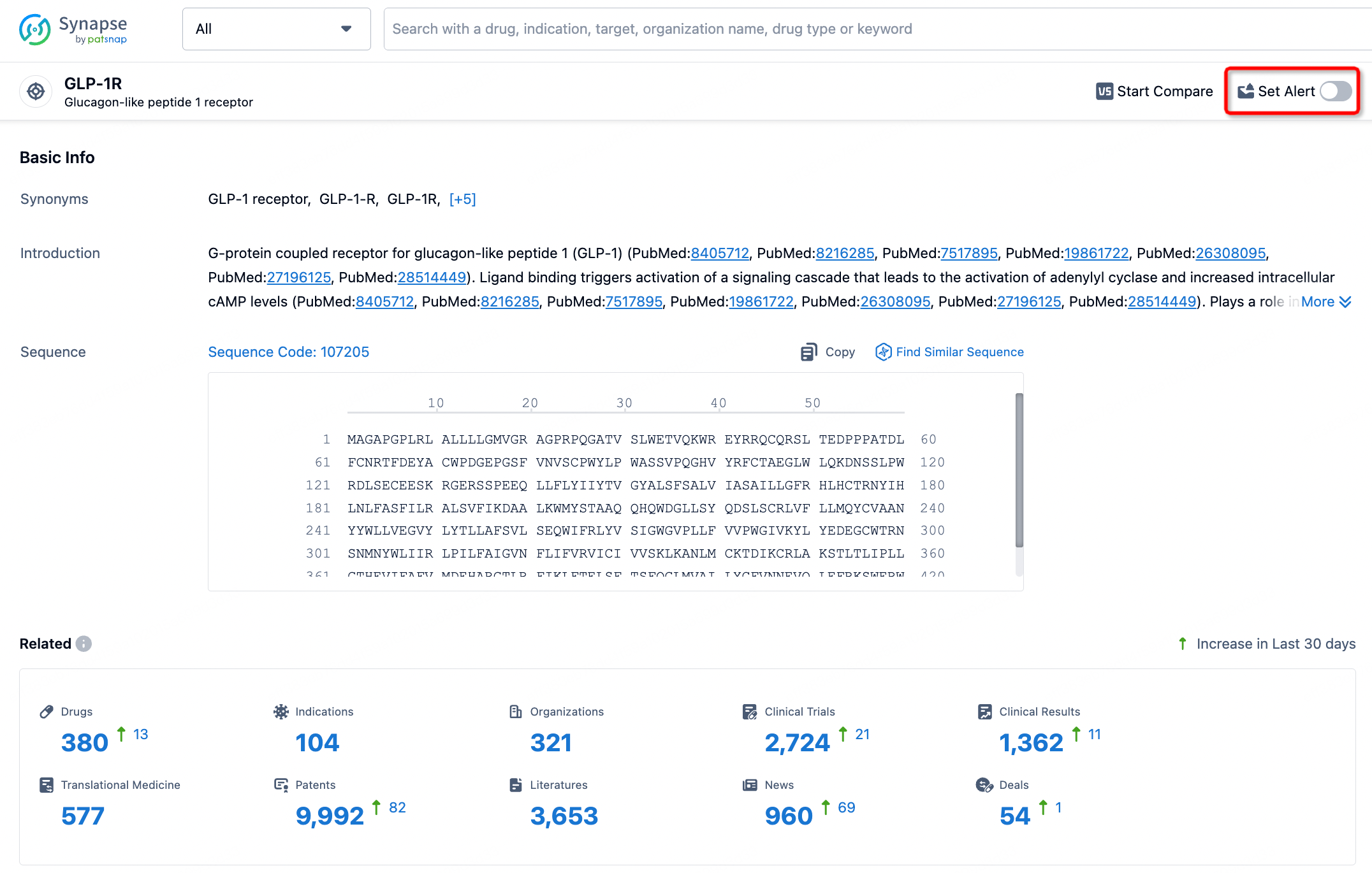
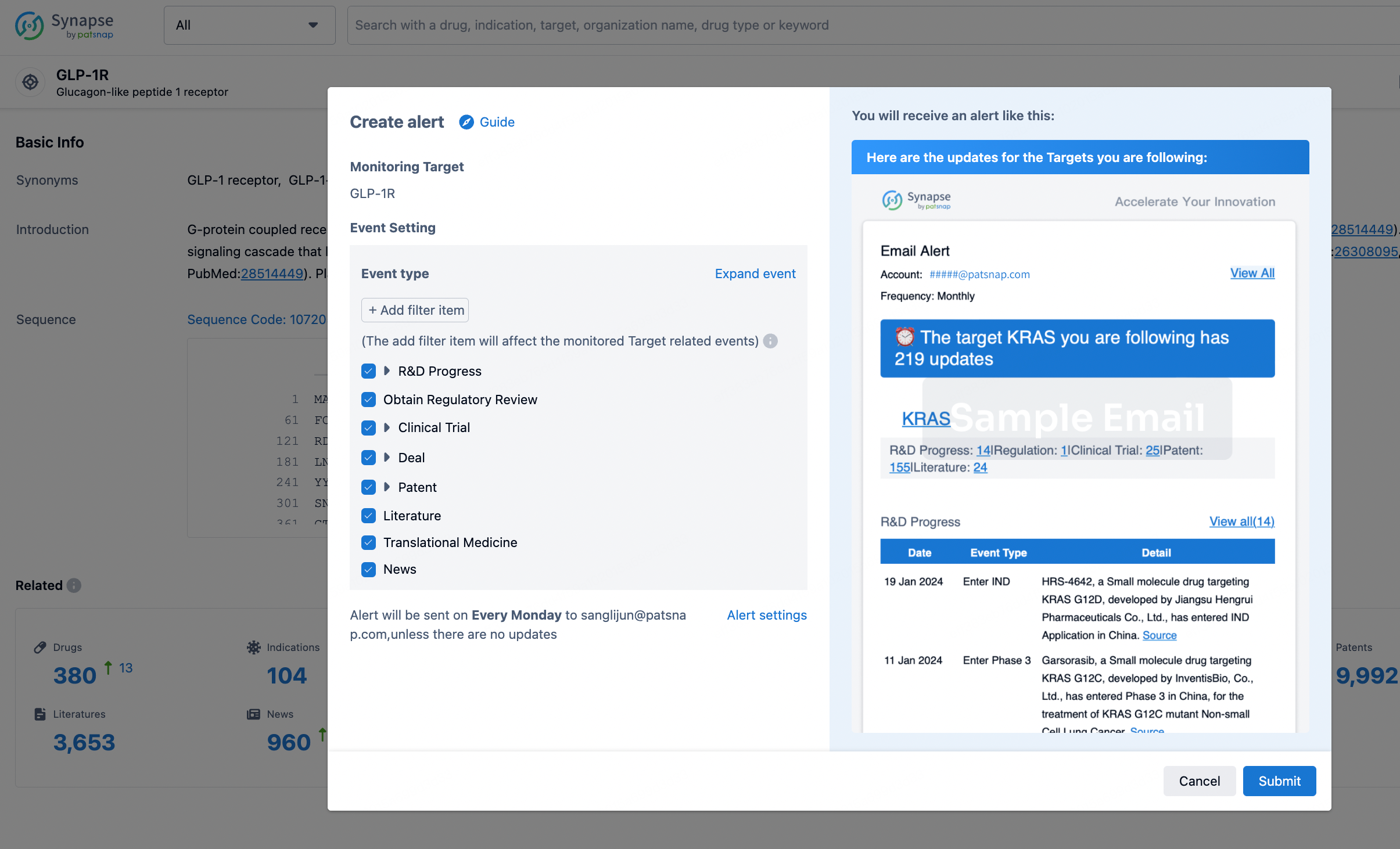
How to obtain the latest development progress of all targets?
In the Synapse database, you can stay updated on the latest research and development advances of all targets. This service is accessible anytime and anywhere, with updates available daily or weekly. Use the "Set Alert" function to stay informed. Click on the image below to embark on a brand new journey of drug discovery!
Hiro LS Researcher
The AI Search Engine Built to Accelerate Biopharma Decisions
Search across billion-scale life sciences data to uncover signals, validate evidence, and act with confidence.
Ask any biopharma research question→
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, Patsnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Discover Synapse Data Servers
Synapse data is now integrated into the PatSnap LS Model Context Protocol (MCP) service. Customize your LLM agent now using our MCP server!