Targeting the Autophagy Pathway as a Potential Novel Therapy for FXS and ASD
Fragile X syndrome (FXS) is the most common genetic cause of intellectual disabilities and a major genetic factor for autism spectrum disorder (ASD). It is characterized by deficits in executive function, learning difficulties, and social anxiety.
A recent study has found that elevated levels of the protein MAP1B result in neuronal autophagy defects closely linked to human ASD. Treating FXS and ASD with rapamycin, a drug that inhibits autophagy, may be a promising potential therapy. As Synapse database indicates, the drug is currently being used for immune System Diseases, neoplasms, congenital Disorders, digestive System Disorders, among many other conditions, but none of the behavioral disorders.
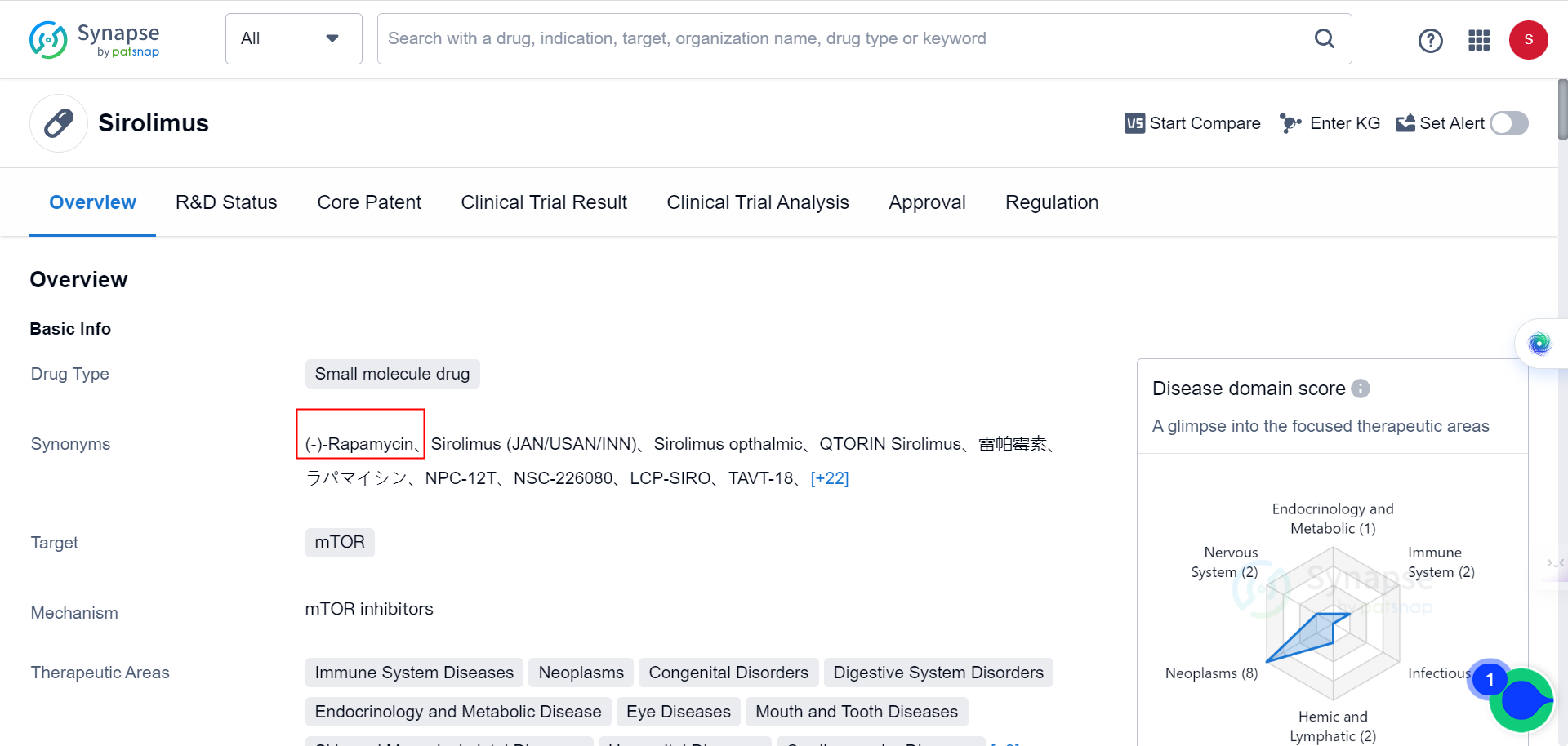
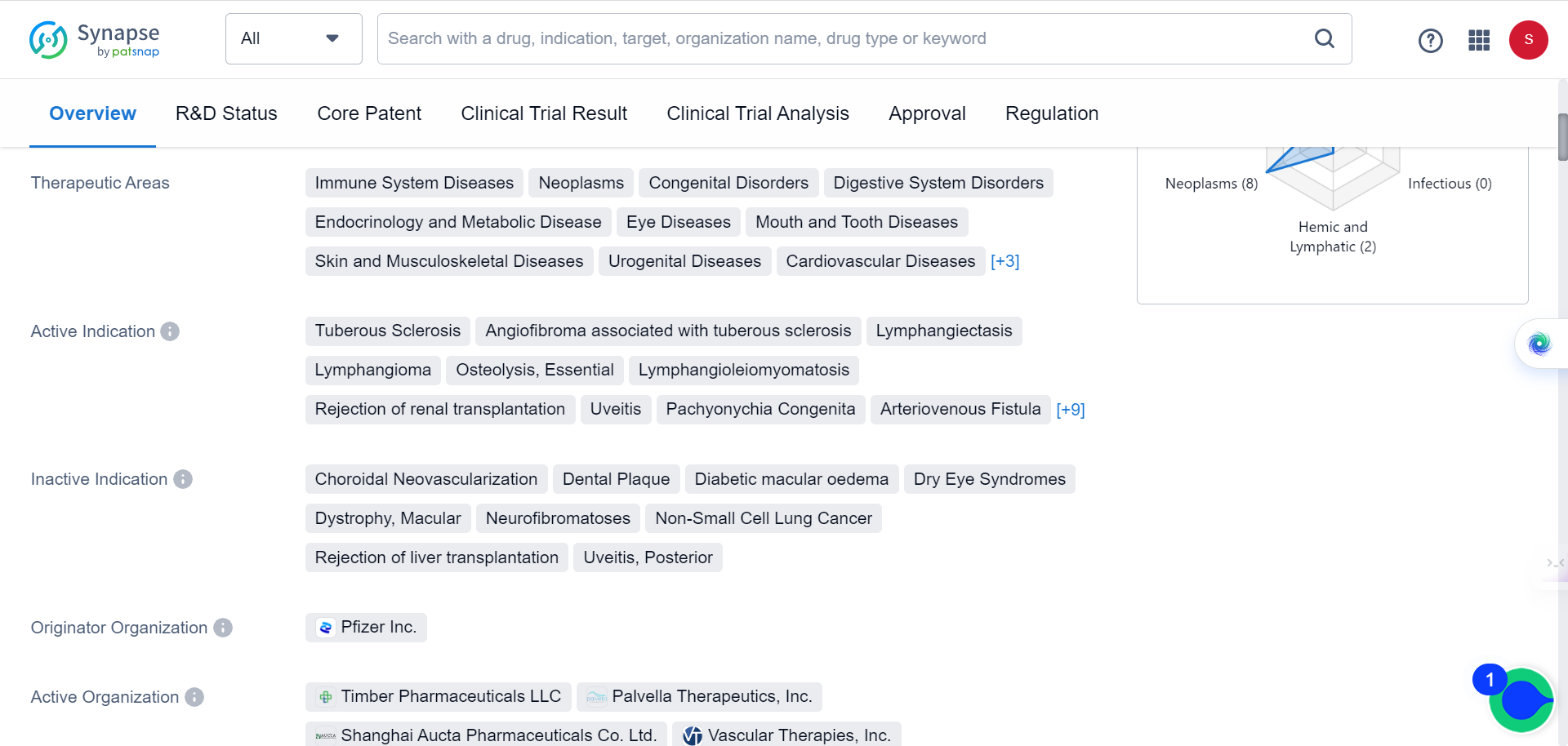
The lack of FMRP
The study observed that, without enough fragile X messenger ribonucleoprotein 1 protein (FMRP), MAP1B levels increase, impairing dendritic development and neuronal activity in humans.
Compared to control cortical neurons infected with LV-shNC, MAP1B expression was significantly higher in cortical neurons infected with LV-shFMR1. Human neurons with elevated MAP1B levels exhibited significantly reduced dendritic complexity, total dendritic length, and increased excitability when compared to control neurons. These might suggest that FMR1 knockdown leads to overexpression of MAP1B, which correlates with dendritic abnormalities and overexcitability in human cortical neurons.
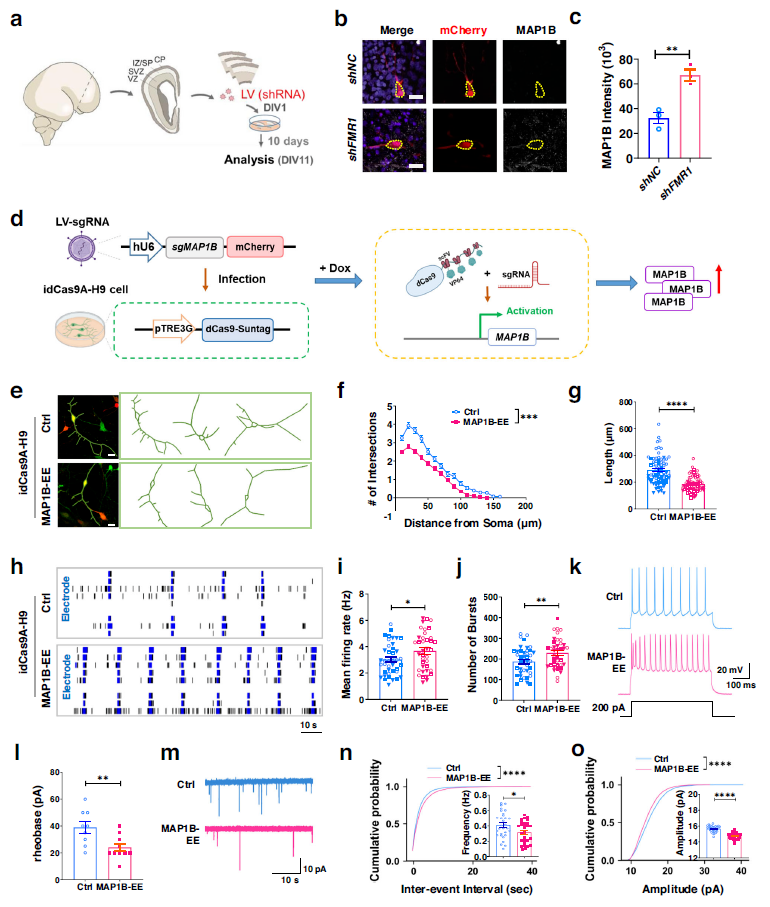
Whole-cell patch clamp recordings were conducted to evaluate the intrinsic excitability of neurons. MAP1B neurons required less injected current to elicit action potentials (lower rheobase current), and the response to current injection showed a higher peak frequency compared to neurons without elevated MAP1B levels, consistent with the increased excitability measured by MEA.
Synaptic transmission in these neurons was further evaluated by recording miniature excitatory postsynaptic currents (mEPSCs). Compared to control neurons, the study found a decrease in the frequency and amplitude of mEPSCs in MAP1B neurons, which may be due to impaired dendritic maturation. Therefore, elevated levels of MAP1B lead to neuronal overexcitability and impaired synaptic transmission in human neurons.
It seems the lack of FMRP leads to an increase in MAP1B levels, which impairs the morphology and electrophysiological maturation of human neurons.
MAP1B and ASD-like social behavior
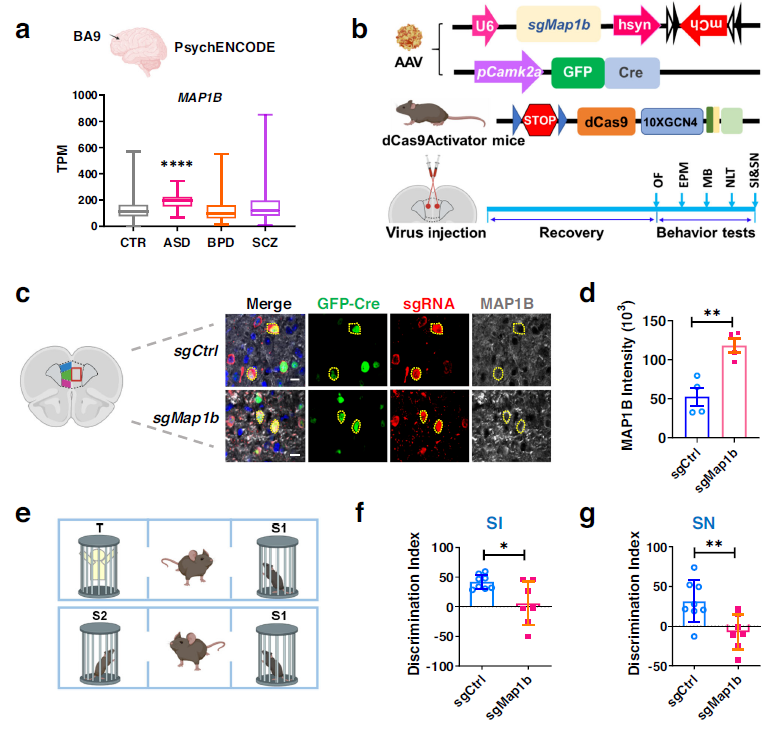
The study also revealed that MAP1B transcript levels were significantly higher in the ASD population compared to the normal population, but not in the BPD or SCZ population. This suggests that MAP1B may be a convergent molecular feature of FXS and ASD. To investigate the potential impact of increased MAP1B expression in the PFC on ASD and FXS-related behaviors in mice, further evaluations were conducted. The results demonstrated that MAP1B in excitatory neurons in the PFC contributes to the social behavior deficits observed in FXS and ASD. Additionally, elevated levels of MAP1B were found to impair neuronal autophagy. These findings suggest a potential mechanism underlying the neurological abnormalities observed in individuals with FXS and ASD and provide insight into potential therapeutic targets for these disorders.
To investigate the molecular mechanisms of MAP1B on dendritic development and behavior, the study used the BioGRID database to identify various autophagy-related proteins that interact with MAP1B. The study first observed that overexpression of MAP1B led to a reduction in LC3, and protein blot analysis showed that the level of LC3-II in neurons overexpressing MAP1B was lower than that in control cells, while the level of LC3-I was unchanged, indicating that MAP1B affects autophagosome formation rather than autophagosome-lysosome fusion. The same result was obtained by inhibiting autophagosome-lysosome fusion with bafilomycin A1 (BafA1).
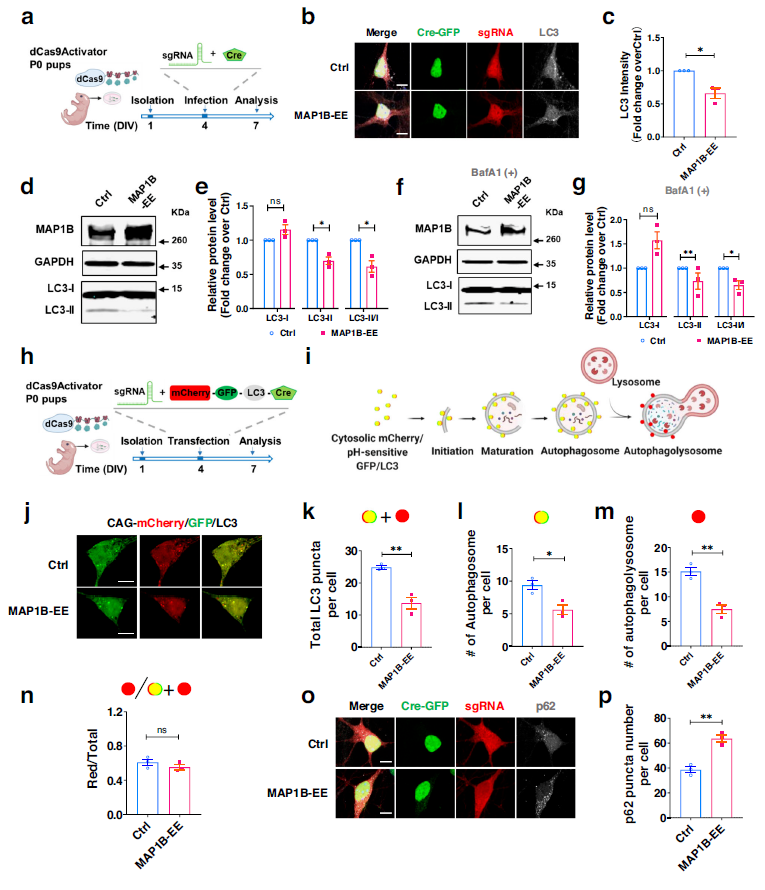
Autophagy defects observed in primary mouse neurons with MAP1B overexpression were due to a reduction in autophagosome formation rather than autophagosome-lysosome fusion. That is, a decrease in autophagosome formation is a common cause of impaired cellular autophagy. Autophagosomes deliver proteins and organelles to lysosomes for degradation through cargo adapter proteins such as p62. When autophagosome formation is reduced, the final degradation step is also reduced, resulting in the accumulation of p62. Compared to the control group, p62 expression increased in neurons overexpressing MAP1B.
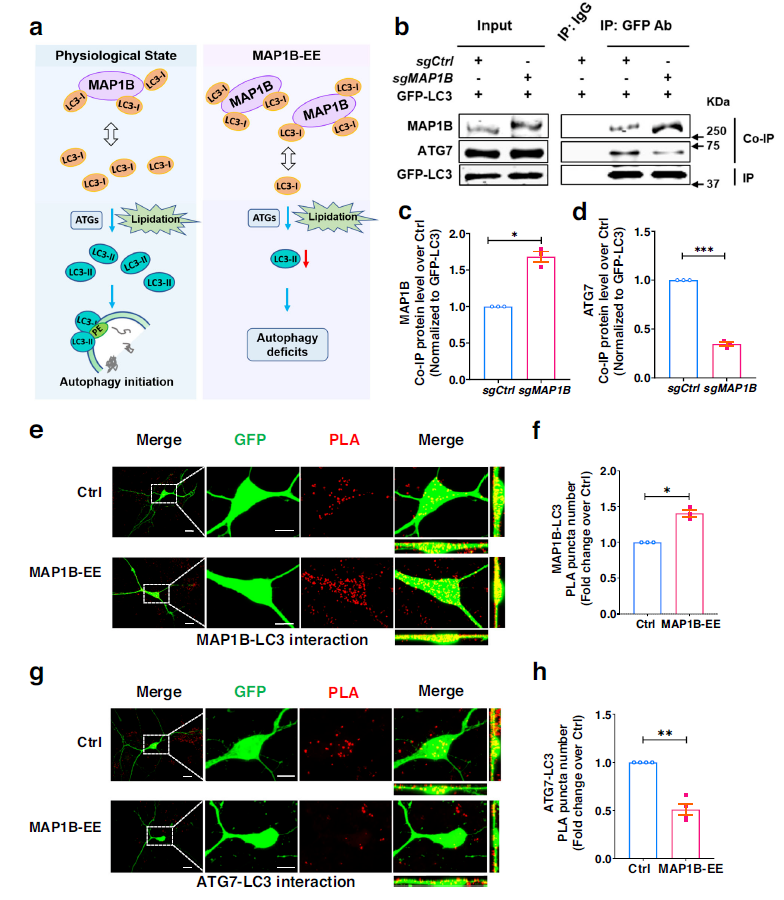
MAP1B and LC3 protein
GFP-LC3 fusion protein from cells overexpressing MAP1B pulled more MAP1B and less ATG7, compared to control neurons, suggesting an increased interaction between MAP1B and LC3, but a decreased interaction between ATG7 and LC3 in cells overexpressing MAP1B. MAP1B overexpression can isolate cell LC3-I, thereby reducing its lipidation and promoting its interaction with MAP1B, rather than with ATG7, which impairs autophagosome formation and leads to autophagy defects.
Furthermore, the study explored whether rapamycin, an autophagy enhancer, could alleviate the deficits caused by MAP1B overexpression. The results demonstrated that rapamycin treatment restored dendritic complexity and total dendritic length in neurons overexpressing MAP1B. Additionally, rapamycin treatment alleviated the social behavior deficits in mice resulting from MAP1B overexpression in the PFC. These findings suggest that the use of autophagy enhancers such as rapamycin may hold therapeutic potential for FXS and ASD, which are associated with MAP1B-related autophagy defects.
This study provides evidence that increased MAP1B levels can negatively impact neuronal development and social behavior through the autophagy pathway, which is associated with FXS and ASD. The findings shed light on the molecular mechanisms underlying these disorders and suggest that rapamycin represents a promising potential therapy for FXS and ASD.

References:
Levine JE, Wang D, Chang Q, Zhao X. Elevated levels of FMRP-target MAP1B impair human and mouse neuronal development and mouse social behaviors via autophagy pathway. Nat Commun. 2023 Jun 26;14(1):3801.



