Request Demo
Last update 17 Jan 2026
SGI-7079
Last update 17 Jan 2026
Overview
Basic Info
Originator Organization |
Active Organization- |
Inactive Organization |
License Organization- |
Drug Highest PhasePendingDiscovery |
First Approval Date- |
Regulation- |


Related
100 Clinical Results associated with SGI-7079
Login to view more data
100 Translational Medicine associated with SGI-7079
Login to view more data
100 Patents (Medical) associated with SGI-7079
Login to view more data
4
Literatures (Medical) associated with SGI-707901 Jun 2015·MOLECULAR CANCER THERAPEUTICS
Correction: Axl Kinase as a Key Target for Oncology: Focus on Small Molecule Inhibitors
Author: Warnault, Pierre ; Feneyrolles, Clemence ; Spenlinhauer, Aurelia ; Fauvel, Benedicte ; Dayde-Cazals, Benedicte ; Cheve, Gwenael ; Guiet, Lea ; Yasri, Aziz
A review.Figure 2 and Supplementary Figure 2 contained incorrect structures for compounds BMS777607, MGCD265, SGI7079, and S49076; the correct structures are given.
01 Feb 2014·LeukemiaQ1 · MEDICINE
The tumor suppressor axis p53/miR-34a regulates Axl expression in B-cell chronic lymphocytic leukemia: implications for therapy in p53-defective CLL patients
Q1 · MEDICINE
Letter
Author: Bearss, D J ; Kay, N E ; Ghosh, A K ; Warner, S L ; Price-Troska, T ; Sinha, S ; Boysen, J ; Viswanatha, D ; Shanafelt, T D
Chronic lymphocytic leukemia (CLL) is an incurable B-cell malignancy and the most common form of leukemia in the Western hemisphere.Recently, we identified a previously undefined receptor tyrosine kinase (RTK), Axl, in CLL B cells,1 as a constitutively active (phosphorylated) RTK that regulates activation of multiple non-receptor cellular kinases including Lyn and phosphatidylinositol 3-kinase/AKT.To this end, the finding that Axl is expressed at variable levels in CLL B cells from CLL patients1 indicates that its regulation is controlled at multiple levels.In addition to its known regulation by multiple transcription factors, post-transcriptional regulation, which has a critical role in modifying and stabilizing protein levels, of Axl remains largely undefined.To explore the possibility of any such regulation in CLL, the entire 3'-untranslated region (UTR) of Axl was analyzed for complementary seed sequences of any known potential miR-binding sites (http://www.microrna.org/microrna/home.do; http://www.targetscan.org), relevant to CLL B cell biol.The most relevant miR-binding target sequence identified was that for the miR-34a (Figure 1a).Of interest, p53 directly regulates the expression of the miR-34 family (miR-34a/b/c), and loss of miR-34 expression is linked to resistance against apoptosis induced by p53-activating agents used in chemotherapies.2 To define that the Axl 3'-UTR is a functional target of miR-34a, we first performed in vitro luciferase reporter gene assays using the reporter construct containing the entire Axl 3'-UTR and increasing amounts of miR-34a mimic or s.c.-miR in HEK293 (human embryonic kidney 293) cells.A dose-dependent reduction of the luciferase activity was observed when the reporter gene and miR-34a mimic were co-transfected (Figure 1b, left panel).As expected, miR-34a did not show any effect on the pMIR-luc-Axl 3'-UTR (-110) reporter gene lacking the miR-34a-binding site (Figure 1b, right panel).We also found that co-transfection of miR-34a mimic with a plasmid DNA expressing the full-length Axl gene reduced Axl expression in a dose-dependent manner (Figure 1c).In addition, enforced introduction of miR-34a mimic in primary CLL B cells reduced endogenous level of Axl (Figure 1d).Taken together, these results suggest that miR-34a targets the Axl 3'-UTR and reduces Axl protein level.During progression of our study,3 two independent groups of investigators have reported a similar complementary binding site for miR-34a in the Axl 3'-UTR;4, 5 however, their findings were limited to established cell lines such that any clin. correlation was less obvious.These initial findings became of more clin. interest as miR-34a, a direct target of p53, is reported to be associated with the adverse outcome in CLL patients.Therefore, we interrogated whether enforced activation of p53 could reduce Axl expression in primary CLL B cells using a DNA-damaging agent, doxorubicin.Results demonstrated accumulation of p53 protein in doxorubicin-treated CLL B cells from all the CLL patients tested (n=14) who had various chromosomal abnormalities detected by fluorescence in situ hybridization (FISH), albeit at variable degrees (Figure 1e).We noted that CLL B cells with deletion of 17p13 or 11q23 (11q23 defects: P6, P7; 17p13 defects: P8, P9) after doxorubicin exposure, accumulated lower levels of p53 and p21, the latter a direct target of p53, relative to the CLL clones with wild-type p53 or the ATM gene, an upstream regulator of p53 function (Figure 1e).Consistent with p53 activation, substantial reduction of Axl expression on doxorubicin-treated CLL B cells was noted including the CLL clones with heterologous deletion of the p53 or ATM gene when compared with the untreated cells (Figure 1f).Next, to establish that it was the increased expression of miR-34a, which reduced Axl expression in doxorubicin-exposed CLL B cells, we measured the levels of mature miR-34a in these CLL B cells.Of note, CLL B cells do not express other members of the miR-34 family: miR-34b/c,8 which recognize the same target binding sequence as does miR-34a.2 An increase of mature miR-34a at variable levels was detected in doxorubicin-treated CLL B cells from different patients (Figure 1g).Interestingly, leukemic B cells with 11q23/17p13 defects also showed variable degrees of miR-34a upregulation in response to doxorubicin (Figure 1g).Multiple studies indicated that p53 could also be activated upon DNA damage independent of ATM via Atr, Chk2 or DNA-PK.9, 10 This raised the possibility that p53 was likely to be activated in 11q23-defected CLL clones independent of ATM upon DNA damage.However, induction of p53 and increase of p21/miR-34a in 17p13-/11q23-defected CLL B cells in response to DNA damage is likely due to the presence of sub-population of cells without the indicated cytogenetic defects and/or these cells have retained one intact wild-type allele.To begin to explore the latter possibility, we sequenced the p53 gene (exons 4-9) in the leukemic B cells from the two 17p13-deleted patients (P8, P9).We found that P8 had a heterozygous splice mutation after exon 4 (GT→TT), although the precise proportion of CLL B cells that had only the heterozygous splice mutation was not known as only 42% of the CLL clone was 17p13 deleted.It is however likely that not all the CLL B cells have this defect as this patient showed activation of the p53 axis upon DNA damage (Figures 1e and g).CLL B cells from P9 harbored splice mutations on the p53 gene before exon 5 (AG→AT and GG→T) after amino acid 153, resulting in a frameshift mutation with a relative abundance of ∼30% in the CLL B cells.This was estimated by relative sequence peak heights at the resp. nucleotide positions.Of note, CLL B cells from P9 had functionally active p53 (Figures 1e and g).An alternative explanation for the differential levels of miR-34a in CLL B cells could be its promoter methylation status.The promoter region containing a p53-binding site and transcription start site of miR-34a gene has been mapped to >30 kb upstream of the mature miR-34a and is within a large (>1.5 kb) CpG island.Indeed, leukemic B cells from CLL patients (P1-P3, P5, P7) who had no 17p13 defects contained both methylated and unmethylated miR-34a promoters (Figure 1h, upper panel).Interestingly, CLL B cells from patients (P10-P14) with no 17p13 defects exhibited predominantly unmethylated miR-34a promoter region except P13, where low-level methylation of the miR-34a promoter was evident (Figure 1h, lower panel).However, CLL B cells from P9 with 17p13 defects had unmethylated miR-34a promoter only, whereas P8 had trace level of methylation and a dominant level of unmethylated miR-34a promoter region (Figure 1h, upper panel).The high level of miR-34a in P9 (Figure 1g) is therefore likely due to both the unmethylated status of miR-34a promoter region and that significant number of the CLL clones had wild-type p53.A relatively low level of miR-34a in P8 (Figure 1g) could be explained by the presence of methylation in the promoter region and slightly higher numbers of 17p13-defective cells.Collectively, these findings suggest that epigenetic regulation of miR-34a expression contributes to the pathobiol. of CLL B cells, independent of p53 status.As fludarabine, a standard chemotherapeutic agent in CLL, is known to induce DNA damage, we were interested in knowing what the impact of this drug has on Axl expression in CLL B cells.Indeed, fludarabine treatment substantially reduced Axl expression on leukemic B cells from CLL patients (n=6) (Figure 1i), likely due to activation of the p53/miR-34a axis in CLL B-cells (Figures 1j and k), suggesting that reduction of Axl expression could be another mechanism by which fludarabine reduces apoptotic resistance and/or induces apoptosis in CLL B cells.Finally, we hypothesized that CLL B cells with 17p13 defects might express higher levels of Axl relative to those with no detectable genetic abnormalities.Indeed, a significantly (P<0.0001) higher levels of Axl expression was detected on CLL B cells from the patients with 17p13 deletion as compared with those with no genetic abnormalities (Figure 2a, left panel).In-depth anal. of the same 17p13 cohort shows that the CLL patients with >75% CLL B cells carrying 17p13 defects express significantly higher levels (P<0.002) of Axl compared with those patients having <75% leukemic B cells with 17p13 defects (Figure 2a, right panel), further indicating that CLL patients with non-functional p53 may express higher levels of Axl.To pursue Axl as a therapeutic target in 17p13-deleted CLL patients, we observed that SGI-7079, a high-affinity Axl inhibitor (Tolero, Lehi, UT, USA), induces massive apoptosis in CLL B cells who had 17p13 deletion in at least 40% of leukemic B cells (n=15) (Figure 2b).Interestingly, when we compared the sensitivity of the leukemic B cells with 17p13 defects vs no genetic abnormalities (normal FISH; n=10) to SGI-7079, no significant difference in LD50 (LD, 50%) doses was detected (Figures 2b and c).Importantly, SGI-7079 induces apoptosis in CLL B cells likely by targeting Axl phosphorylation (Figure 2d).However, we cannot completely rule out off-target effects of SGI-7079 on other RTKs.Collectively, these results suggest that Axl is a targetable RTK in CLL and provides a future therapeutic option for very 'high-risk' CLL patients with non-functional p53.In summary, we found that p53 activation neg. regulates Axl expression via upregulating miR-34a in CLL B cells with functional p53 (Figure 2e).Although this study found an inverse link between p53 inactivation and Axl expression in CLL B cells, we are aware that Axl can also be regulated by other mechanisms including multiple transcription factors and/or epigenetic modulations.However, our study uniquely adds to the growing body of literature regarding Axl regulation in human malignancies and suggests that p53 inactivation stabilizes Axl protein levels in CLL.Thus, Axl inhibition in CLL should be considered and evaluated as a potential therapy (Figure 2e).
01 Jan 2013·Clinical cancer research : an official journal of the American Association for Cancer ResearchQ1 · MEDICINE
An Epithelial–Mesenchymal Transition Gene Signature Predicts Resistance to EGFR and PI3K Inhibitors and Identifies Axl as a Therapeutic Target for Overcoming EGFR Inhibitor Resistance
Q1 · MEDICINE
Article
Author: George R. Blumenschein ; Lixia Diao ; Gordon B. Mills ; Lauren Averett Byers ; Varsha Gandhi ; Robert J.G. Cardnell ; Steven L. Warner ; Ignacio I. Wistuba ; Roy S. Herbst ; Waun K. Hong ; J. Jack Lee ; K. Kian Ang ; John N. Weinstein ; Hai Tran ; Jason M. Foulks ; Luc Girard ; Uma Giri ; Pierre Saintigny ; Youhong Fan ; John V. Heymach ; Jing Wang ; David J. Bearss ; Kevin R. Coombes ; Nancy Krett ; Steven B. Kanner ; Edward S. Kim ; Praveen K. Tumula ; Michael Peyton ; Steven T. Rosen ; Li Shen ; Scott M. Lippman ; John D. Minna ; Monique B. Nilsson ; Jayanthi Gudikote
Abstract:
Purpose: Epithelial–mesenchymal transition (EMT) has been associated with metastatic spread and EGF receptor (EGFR) inhibitor resistance. We developed and validated a robust 76-gene EMT signature using gene expression profiles from four platforms using non–small cell lung carcinoma (NSCLC) cell lines and patients treated in the Biomarker-Integrated Approaches of Targeted Therapy for Lung Cancer Elimination (BATTLE) study.Experimental Design: We conducted an integrated gene expression, proteomic, and drug response analysis using cell lines and tumors from patients with NSCLC. A 76-gene EMT signature was developed and validated using gene expression profiles from four microarray platforms of NSCLC cell lines and patients treated in the BATTLE study, and potential therapeutic targets associated with EMT were identified.Results: Compared with epithelial cells, mesenchymal cells showed significantly greater resistance to EGFR and PI3K/Akt pathway inhibitors, independent of EGFR mutation status, but more sensitivity to certain chemotherapies. Mesenchymal cells also expressed increased levels of the receptor tyrosine kinase Axl and showed a trend toward greater sensitivity to the Axl inhibitor SGI-7079, whereas the combination of SGI-7079 with erlotinib reversed erlotinib resistance in mesenchymal lines expressing Axl and in a xenograft model of mesenchymal NSCLC. In patients with NSCLC, the EMT signature predicted 8-week disease control in patients receiving erlotinib but not other therapies.Conclusion: We have developed a robust EMT signature that predicts resistance to EGFR and PI3K/Akt inhibitors, highlights different patterns of drug responsiveness for epithelial and mesenchymal cells, and identifies Axl as a potential therapeutic target for overcoming EGFR inhibitor resistance associated with the mesenchymal phenotype. Clin Cancer Res; 19(1); 279–90. ©2012 AACR.
100 Deals associated with SGI-7079
Login to view more data
R&D Status
10 top R&D records. to view more data
Login
| Indication | Highest Phase | Country/Location | Organization | Date |
|---|---|---|---|---|
| Neoplasms | Discovery | United States | 22 Oct 2012 | |
| Neoplasms | Discovery | United States | 22 Oct 2012 |
Login to view more data
Clinical Result
Clinical Result
Indication
Phase
Evaluation
View All Results
| Study | Phase | Population | Analyzed Enrollment | Group | Results | Evaluation | Publication Date |
|---|
No Data | |||||||
Login to view more data
Translational Medicine
Boost your research with our translational medicine data.
login
or

Deal
Boost your decision using our deal data.
login
or

Core Patent
Boost your research with our Core Patent data.
login
or
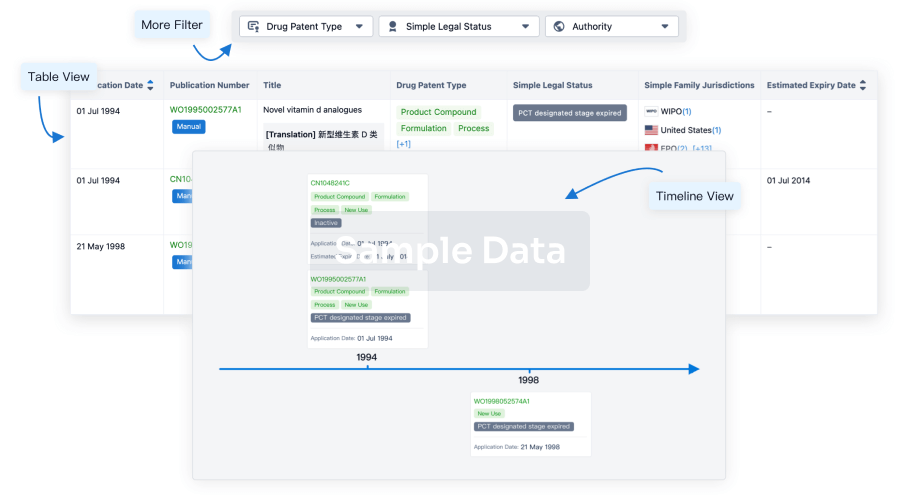
Clinical Trial
Identify the latest clinical trials across global registries.
login
or

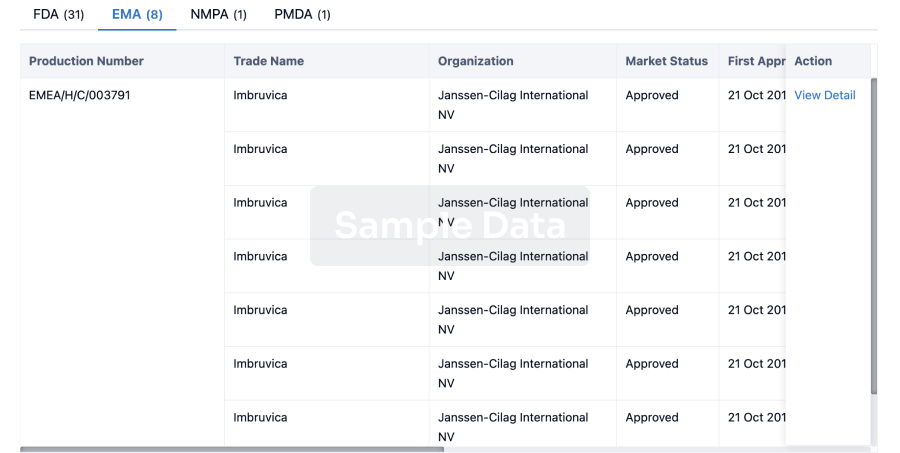
Approval
Accelerate your research with the latest regulatory approval information.
login
or
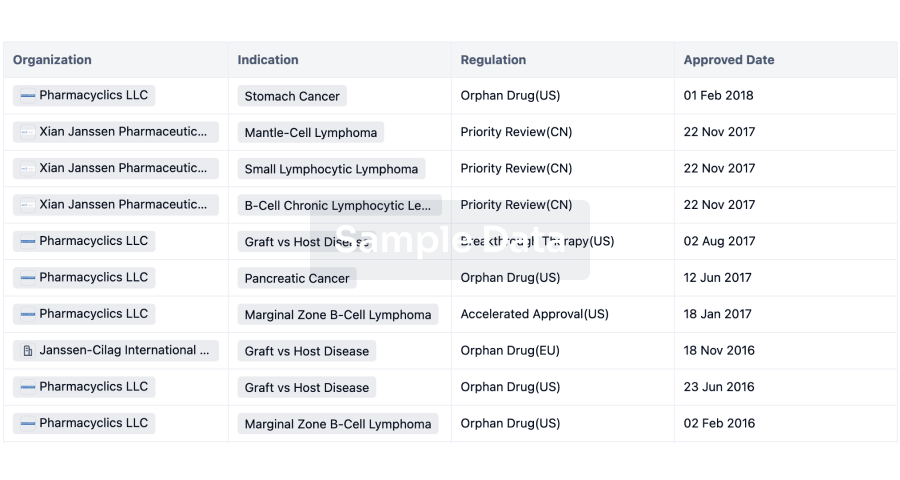
Regulation
Understand key drug designations in just a few clicks with Synapse.
login
or
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.
Bio
Bio Sequences Search & Analysis
Sign up for free
Chemical
Chemical Structures Search & Analysis
Sign up for free