CDK2, or cyclin-dependent kinase 2, is a crucial protein involved in cell cycle regulation in the human body. It plays a pivotal role in controlling the progression of cells from the G1 phase to the S phase, allowing DNA replication and cell division to occur. CDK2 forms complexes with cyclins, which activate its kinase activity, leading to the phosphorylation of various target proteins involved in cell cycle progression.
The CDK2/cyclin E complex plays a crucial role in the phosphorylation of retinoblastoma (Rb) protein and the transition from G1/S phase. The CDK2/cyclin A complex is significantly involved in DNA synthesis during the S phase and the activation of CDK1/cyclin B during the transition from G2/M phase. PF-07104091(19) is an orally available CDK2 inhibitor with a potential antitumor activity. In human ovarian cancer cell models(OVCAR3, OV5392), compound 19(25, 75 and 175mg/kg, twice daily, po) can induce tumor reduction in a dose-dependent manner. Furthermore, the combined administration of 19 and the frontline drug Palbociclib demonstrated a synergistic effect in the model of breast cancer cells. In 2020, Pfizer initiated a Phase I/II clinical trial of compound 19 to assess its safety and tolerance and to test the maximum tolerated dose of compound 19 as a single therapy in patients with small cell lung cancer, non-small cell lung cancer, ovarian cancer or breast cancer(NCT04553133).
Dysregulation of CDK2 activity has been implicated in numerous diseases, including cancer, making it an attractive target for therapeutic interventions aimed at controlling cell proliferation and promoting healthy cell cycle prThe target CDK2 in the pharmaceutical industry has attracted the attention of several companies, including Pfizer Inc., Cyclacel Pharmaceuticals, Inc., Otsuka Holdings Co., Ltd., and Tiziana Life Sciences Ltd. These companies are actively involved in the research and development of drugs targeting CDK2. Indications such as non-small cell lung cancer, ovarian cancer, and triple negative breast cancer have seen the approval of drugs targeting CDK2. The most rapidly progressing drug types under the target CDK2 are small molecule drugs, indicating intense competition in this area. The European Union and the United States are leading in terms of development, with China also making progress. Overall, the competitive landscape for target CDK2 is dynamic, and future development in this area holds significant potential.ogression.
How do they work?CDK2 inhibitors are a type of drugs that specifically target and inhibit the activity of cyclin-dependent kinase 2 (CDK2). CDK2 is a protein kinase that plays a crucial role in regulating the cell cycle, specifically the transition from the G1 phase to the S phase. By inhibiting CDK2, these inhibitors can disrupt the cell cycle progression and prevent the proliferation of cancer cells.
From a biomedical perspective, CDK2 inhibitors are of great interest in the field of cancer research and treatment. Cancer cells often exhibit uncontrolled cell division and proliferation, and CDK2 is frequently overactive in these cells. By blocking CDK2 activity, CDK2 inhibitors can potentially halt the growth of cancer cells and serve as a targeted therapy for various types of cancer.
It's important to note that CDK2 inhibitors are still under investigation and development, and their clinical use is currently limited to clinical trials. These inhibitors hold promise as potential anticancer agents, but further research is needed to determine their efficacy, safety, and optimal usage in cancer treatment.
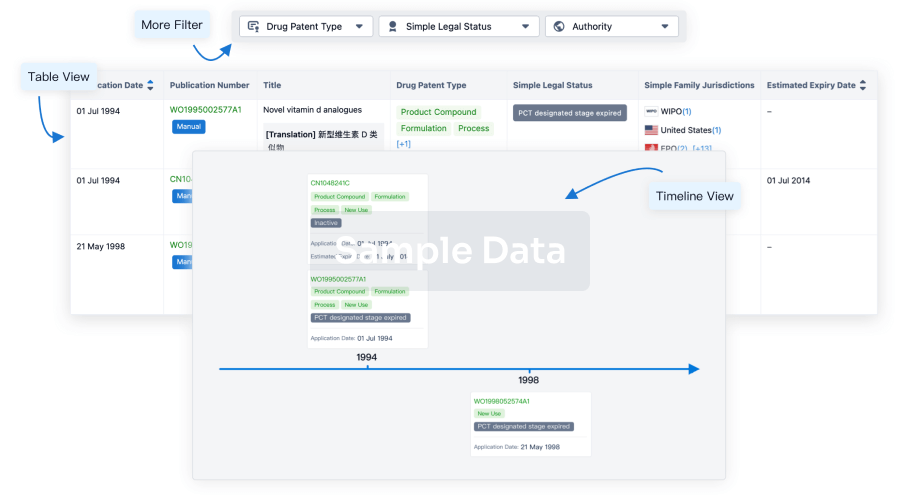
List of CDK2 InhibitorsThe currently marketed CDK2 inhibitors include:
AT-7519Milciclib maleatePF-06873600PF-07104091SeliciclibARTS-021BLU-222FadraciclibINX-315DinaciclibFor more information, please click on the image below.
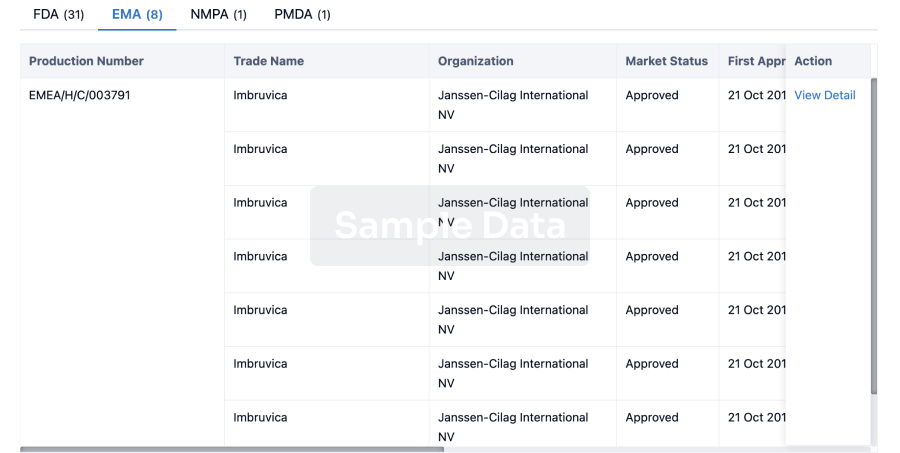
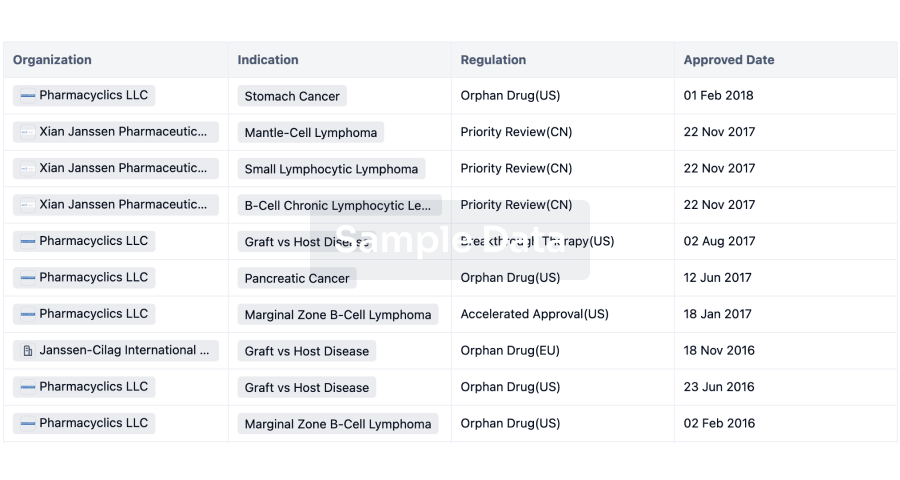
What are CDK2 inhibitors used for?CDK2 inhibitors are under investigation forIndications such as non-small cell lung cancer, ovarian cancer, and triple negative breast cancer.For more information, please click on the image below to log in and search.
How to obtain the latest development progress of CDK2 inhibitors?In the Synapse database, you can keep abreast of the latest research and development advances of CDK2 inhibitors anywhere and anytime, daily or weekly, through the "Set Alert" function. Click on the image below to embark on a brand new journey of drug discovery!