Request Demo
Last update 08 May 2025
P4HTM
Last update 08 May 2025
Basic Info
Synonyms EGLN4, FLJ20262, HIF-PH4 + [11] |
Introduction Catalyzes the post-translational formation of 4-hydroxyproline in hypoxia-inducible factor (HIF) alpha proteins. Hydroxylates HIF1A at 'Pro-402' and 'Pro-564'. May function as a cellular oxygen sensor and, under normoxic conditions, may target HIF through the hydroxylation for proteasomal degradation via the von Hippel-Lindau ubiquitination complex. |
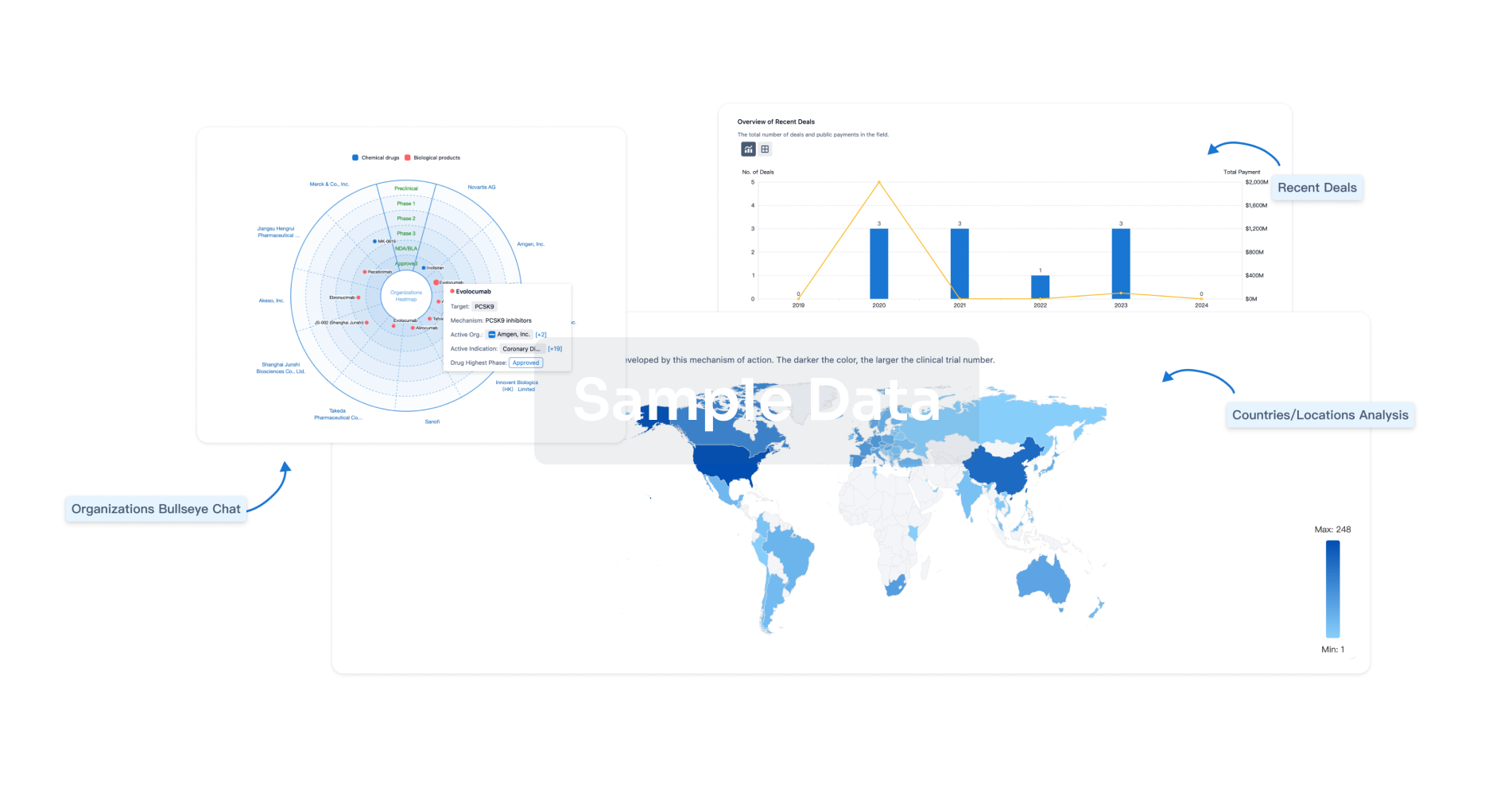
Analysis
Perform a panoramic analysis of this field.
login
or
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.
Bio
Bio Sequences Search & Analysis
Sign up for free
Chemical
Chemical Structures Search & Analysis
Sign up for free