Request Demo
Last update 07 Oct 2024
BMF
Last update 07 Oct 2024
Basic Info
Related Targets |
Related
100 Clinical Results associated with BMF
Login to view more data
100 Translational Medicine associated with BMF
Login to view more data
0 Patents (Medical) associated with BMF
Login to view more data
310
Literatures (Medical) associated with BMF01 Dec 2024·TRANSFUSION AND APHERESIS SCIENCE
Does immunohistochemical staining predict mobilization success in multiple myeloma patients?
Article
Author: Demir, Derya ; Karadag, Fatma Keklik ; Güneş, Ajda ; Soyer, Nur ; Arslan, Aysenur ; Sahin, Fahri ; Vural, Filiz ; Töbü, Mahmut ; Saydam, Guray ; Bozer, Denis ; Aysin, Murat
BACKGROUND:
So many risk factors for mobilization failure have been described so far. We aimed to identify the risk factors and search the possible effects of bone marrow fibrosis (BMF), CD56, c-myc, and cyclinD1 expression on mobilization.
METHODS:
We evaluated 189 patients with MM who were admitted for stem cell mobilization before autologous stem cell transplantation (ASCT) between 2015 and June 2021. Clinical, laboratory, treatment features, and survival outcomes were compared in patients who were successfully mobilized and who were not.
RESULTS:
Mobilization failure rate was 11.1 % (21) in our study group. Male gender, mobilization with only G-CSF, history of previous ASCT, lenalidomide exposure, and 2 lines of chemotherapy before stem cell mobilization were observed more commonly in mobilization failure group. There is no relationship between mobilization failure and BMF, CD56, c-myc, and cyclin D1 expression status in patients who received either only G-CSF or G-CSF+ chemotherapy for mobilization. Overall survival (OS) was not different in groups of patients who were successfully mobilized and who were not. Neutrophil engraftment was faster in patients who were transfused > 5 × 106/kg stem cells (p = 0.015). ECOG performance status (p = 0.004), c-myc expression (p = 0.005), lenalidomide therapy before mobilization (p = 0.032), and mobilization with G-CSF+chemotherapy was found to be predictive factors for OS.
CONCLUSION:
Even though we could not find any predictive value of CD56, c-myc, and cyclin D1 expression on mobilization, c-myc was found to be associated with low OS. Further studies with large and homogenous study population would be more informative.
01 Nov 2024·TOXICOLOGY
Proteasome inhibition induces apoptosis through simultaneous inactivation of MCL-1/BCL-XL by NOXA independent of CHOP and JNK pathways
Article
Author: Chen, Wenjuan ; Dong, Na ; Fu, Rongrong ; Sun, Mengning ; Yang, Qinglan ; Sun, Yi ; Gao, Hui ; Li, Li
Proteasome inhibitors have been employed in the treatment of relapsed multiple myeloma and mantle cell lymphoma. The observed toxicity caused by proteasome inhibitors is a universal phenotype in numerous cancer cells with different sensitivity. In this study, we investigate the conserved mechanisms underlying the toxicity of the proteasome inhibitor bortezomib using gene editing approaches. Our findings utilizing different caspase knocking out cells reveal that bortezomib induces classic intrinsic apoptosis by activating caspase-9 and caspase-3/7, leading to pore-forming protein GSDME cleavage and subsequent lytic cell death or called secondary necrosis, a phenotype also observed in many apoptosis triggers like TNFα plus CHX, DTT and tunicamycin treatment in HeLa cells. Furthermore, through knocking out of nearly all BH3-only proteins including BIM, BAD, BID, BMF and PUMA, we demonstrate that NOXA is the sole BH3-only protein responsible for bortezomib-induced apoptosis. Of note, NOXA is well known for selectively binding to MCL-1 and A1, but our studies utilizing different BH3 mimetics as well as immunoprecipitation assays indicate that, except for the constitutive interaction of NOXA with MCL-1, the accumulation of NOXA after bortezomib treatment allows it to interact with BCL-XL, then simultaneous relieving suppression on apoptosis by both anti-apoptotic proteins BCL-XL and MCL-1. In addition, though bortezomib-induced significant ER stress and JNK activation were observed in the study, further genetic depletion experiments prove that bortezomib-induced apoptosis occurs independently of ER stress-related apoptosis factor CHOP and JNK. In summary, these results provide a solid conclusion about the critical role of NOXA in inactivation of BCL-XL except MCL-1 in bortezomib-induced apoptosis.
01 Oct 2024·Biomaterials advances
Matrix stiffening facilitates stemness of liver cancer stem cells by YAP activation and BMF inhibition
Article
Author: Ma, Di ; Luo, Qing ; Song, Guanbin
Matrix stiffening is one of the major risk factors for hepatocellular carcinoma (HCC) and drives tumor progression. The extracellular matrix (ECM) stiffness of HCC displays mechanical heterogeneity, with stiffness increasing from the core to the invasive frontier. The distribution of liver cancer stem cells (CSCs) is related to this mechanical property. However, it is not sufficiently understood how heterogeneous matrix stiffness regulates the stemness of CSCs. In this study, we developed an adjustable gelatin/alginate hydrogel to investigate the effect of various matrix stiffnesses on CSC stemness under three-dimensional culture conditions. Gelatin/alginate hydrogel with the stiffness of soft (5 kPa), medium (16 kPa), and stiff (81 kPa) were prepared by altering the concentration of calcium ions. It was found that a stiffer matrix promoted stemness-associated gene expression, reduced drug sensitivity, enhanced sphere-forming and clonogenic ability, and tumorigenic potential. Mechanistically, matrix stiffening facilitates CSC stemness by increasing Yes-associated protein (YAP) activity and inhibiting Bcl-2 modifying factor (BMF) expression. Knockdown of YAP or overexpression of BMF significantly attenuated matrix stiffening-induced stemness, suggesting the involvement of YAP and BMF in this process. Together, our results unravel the regulatory mechanism of heterogeneous matrix stiffness on CSC stemness and also provide a novel therapeutic strategy for eradicating CSCs and improving the efficiency of HCC treatment.
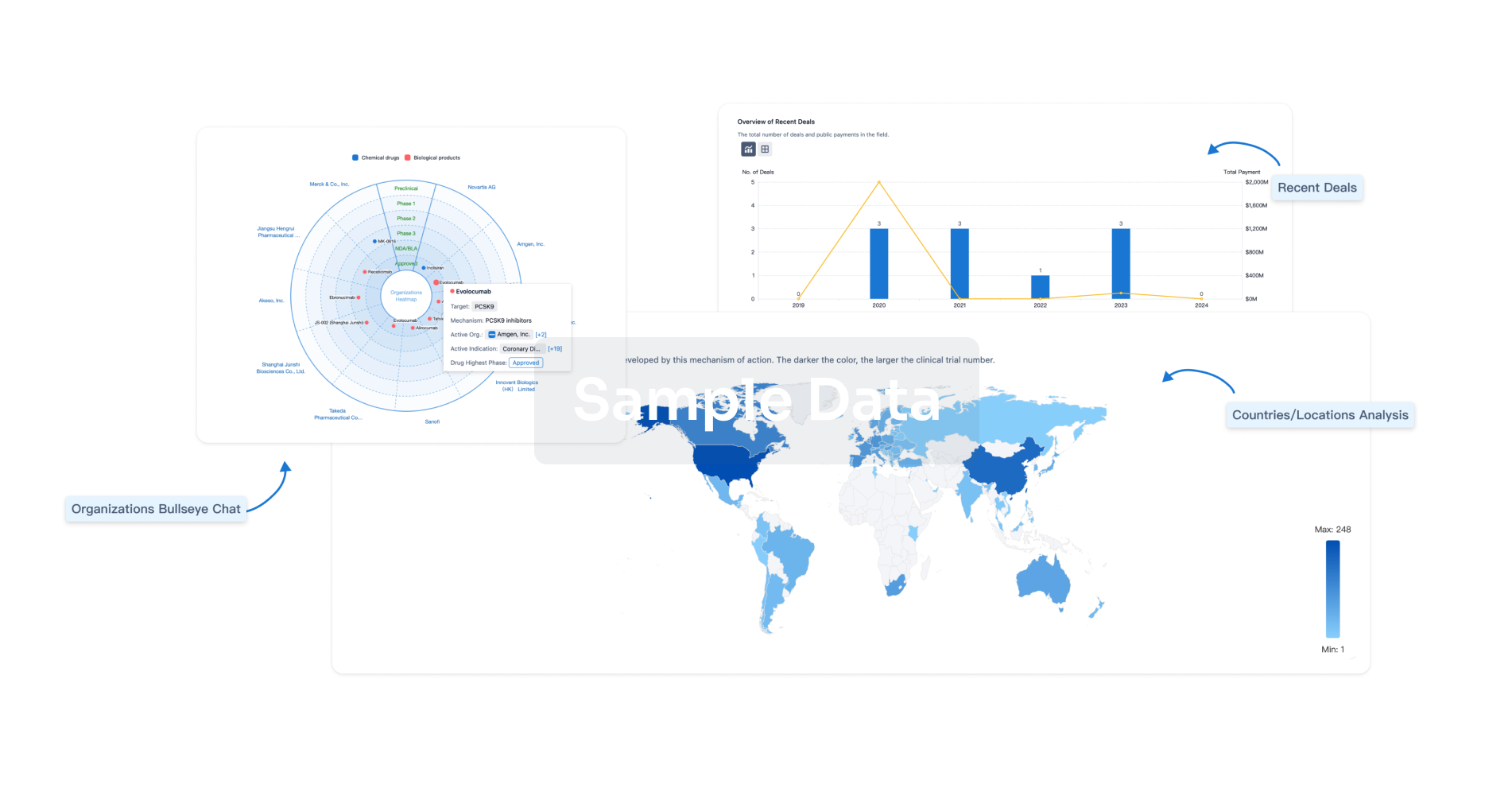
Analysis
Perform a panoramic analysis of this field.
login
or
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.
Bio
Bio Sequences Search & Analysis
Sign up for free
Chemical
Chemical Structures Search & Analysis
Sign up for free