Request Demo
What is Lucerastat used for?
28 June 2024
Introduction to Lucerastat
Lucerastat is a small molecule drug that has gained considerable attention in recent years for its potential in treating rare genetic disorders. Developed by Idorsia Pharmaceuticals, Lucerastat specifically targets sphingolipid metabolism, offering a novel therapeutic approach. Sphingolipids are a class of lipids that play a crucial role in cell membrane structure and function. Abnormalities in sphingolipid metabolism are implicated in various lysosomal storage diseases, including Gaucher disease and Fabry disease. These diseases are characterized by the accumulation of lipids in cells due to defective enzymes, leading to a range of debilitating symptoms and complications.
The drug is currently undergoing clinical trials to evaluate its efficacy and safety in patients with Fabry disease, a genetic disorder caused by mutations in the GLA gene. These mutations lead to a deficiency in the enzyme alpha-galactosidase A, resulting in the accumulation of a specific sphingolipid called globotriaosylceramide (Gb3) in tissues throughout the body. The buildup of Gb3 leads to progressive kidney damage, heart disease, and other systemic complications. The promising results from preclinical studies have paved the way for current Phase III clinical trials, aimed at substantiating its therapeutic potential.
Lucerastat Mechanism of Action
Lucerastat operates through a mechanism of action that targets the biosynthesis pathway of glycosphingolipids (GSLs). Unlike enzyme replacement therapies (ERTs) currently used to treat lysosomal storage diseases, Lucerastat works by inhibiting the enzyme glucosylceramide synthase (GCS). GCS is responsible for the first step in the synthesis of glycosphingolipids. By suppressing GCS activity, Lucerastat reduces the accumulation of harmful sphingolipids in cells.
This mechanism represents a substrate reduction therapy (SRT) approach, aimed at decreasing the production of the substrates that accumulate due to enzyme deficiencies. In the context of Fabry disease, reducing the synthesis of glycosphingolipids helps to lower the levels of Gb3 and its derivative, globotriaosylsphingosine (Lyso-Gb3), thereby mitigating the disease's pathological manifestations. The advantage of this approach is that it targets the root cause of the lipid accumulation rather than just addressing the symptoms.
What is the indication of Lucerastat?
Lucerastat is primarily being developed for the treatment of Fabry disease. Fabry disease is an X-linked lysosomal storage disorder that affects multiple organ systems, including the kidneys, heart, and skin. Patients with Fabry disease typically present with a range of symptoms such as acroparesthesias (painful burning sensations in the hands and feet), angiokeratomas (small, dark red spots on the skin), corneal opacity, renal dysfunction, and cardiovascular complications. The disease is progressive, and without adequate treatment, it can lead to severe and life-threatening conditions, including end-stage renal disease and heart failure.
Currently, the standard treatment for Fabry disease involves enzyme replacement therapy (ERT), where patients receive intravenous infusions of recombinant alpha-galactosidase A to break down the accumulated Gb3. While ERT has been shown to be effective in reducing Gb3 levels and alleviating some symptoms, it has limitations. ERT requires bi-weekly infusions, is associated with significant costs, and some patients develop antibodies against the recombinant enzyme, which can reduce its efficacy.
Lucerastat offers a promising alternative, especially for patients who are either intolerant to or unresponsive to ERT. The oral administration of Lucerastat presents a more convenient option, potentially improving patient compliance and quality of life. Moreover, as a substrate reduction therapy, Lucerastat could be used in conjunction with other treatments, providing a multi-faceted approach to managing Fabry disease.
In conclusion, Lucerastat is an innovative drug that targets sphingolipid metabolism, offering a new therapeutic strategy for treating Fabry disease. Its mechanism of action as a glucosylceramide synthase inhibitor marks a significant departure from the traditional enzyme replacement therapies, aiming to address the root cause of the lipid accumulation. As clinical trials progress, Lucerastat holds the potential to become a vital tool in the arsenal against Fabry disease, offering hope to patients and families affected by this challenging genetic disorder.
Lucerastat is a small molecule drug that has gained considerable attention in recent years for its potential in treating rare genetic disorders. Developed by Idorsia Pharmaceuticals, Lucerastat specifically targets sphingolipid metabolism, offering a novel therapeutic approach. Sphingolipids are a class of lipids that play a crucial role in cell membrane structure and function. Abnormalities in sphingolipid metabolism are implicated in various lysosomal storage diseases, including Gaucher disease and Fabry disease. These diseases are characterized by the accumulation of lipids in cells due to defective enzymes, leading to a range of debilitating symptoms and complications.
The drug is currently undergoing clinical trials to evaluate its efficacy and safety in patients with Fabry disease, a genetic disorder caused by mutations in the GLA gene. These mutations lead to a deficiency in the enzyme alpha-galactosidase A, resulting in the accumulation of a specific sphingolipid called globotriaosylceramide (Gb3) in tissues throughout the body. The buildup of Gb3 leads to progressive kidney damage, heart disease, and other systemic complications. The promising results from preclinical studies have paved the way for current Phase III clinical trials, aimed at substantiating its therapeutic potential.
Lucerastat Mechanism of Action
Lucerastat operates through a mechanism of action that targets the biosynthesis pathway of glycosphingolipids (GSLs). Unlike enzyme replacement therapies (ERTs) currently used to treat lysosomal storage diseases, Lucerastat works by inhibiting the enzyme glucosylceramide synthase (GCS). GCS is responsible for the first step in the synthesis of glycosphingolipids. By suppressing GCS activity, Lucerastat reduces the accumulation of harmful sphingolipids in cells.
This mechanism represents a substrate reduction therapy (SRT) approach, aimed at decreasing the production of the substrates that accumulate due to enzyme deficiencies. In the context of Fabry disease, reducing the synthesis of glycosphingolipids helps to lower the levels of Gb3 and its derivative, globotriaosylsphingosine (Lyso-Gb3), thereby mitigating the disease's pathological manifestations. The advantage of this approach is that it targets the root cause of the lipid accumulation rather than just addressing the symptoms.
What is the indication of Lucerastat?
Lucerastat is primarily being developed for the treatment of Fabry disease. Fabry disease is an X-linked lysosomal storage disorder that affects multiple organ systems, including the kidneys, heart, and skin. Patients with Fabry disease typically present with a range of symptoms such as acroparesthesias (painful burning sensations in the hands and feet), angiokeratomas (small, dark red spots on the skin), corneal opacity, renal dysfunction, and cardiovascular complications. The disease is progressive, and without adequate treatment, it can lead to severe and life-threatening conditions, including end-stage renal disease and heart failure.
Currently, the standard treatment for Fabry disease involves enzyme replacement therapy (ERT), where patients receive intravenous infusions of recombinant alpha-galactosidase A to break down the accumulated Gb3. While ERT has been shown to be effective in reducing Gb3 levels and alleviating some symptoms, it has limitations. ERT requires bi-weekly infusions, is associated with significant costs, and some patients develop antibodies against the recombinant enzyme, which can reduce its efficacy.
Lucerastat offers a promising alternative, especially for patients who are either intolerant to or unresponsive to ERT. The oral administration of Lucerastat presents a more convenient option, potentially improving patient compliance and quality of life. Moreover, as a substrate reduction therapy, Lucerastat could be used in conjunction with other treatments, providing a multi-faceted approach to managing Fabry disease.
In conclusion, Lucerastat is an innovative drug that targets sphingolipid metabolism, offering a new therapeutic strategy for treating Fabry disease. Its mechanism of action as a glucosylceramide synthase inhibitor marks a significant departure from the traditional enzyme replacement therapies, aiming to address the root cause of the lipid accumulation. As clinical trials progress, Lucerastat holds the potential to become a vital tool in the arsenal against Fabry disease, offering hope to patients and families affected by this challenging genetic disorder.
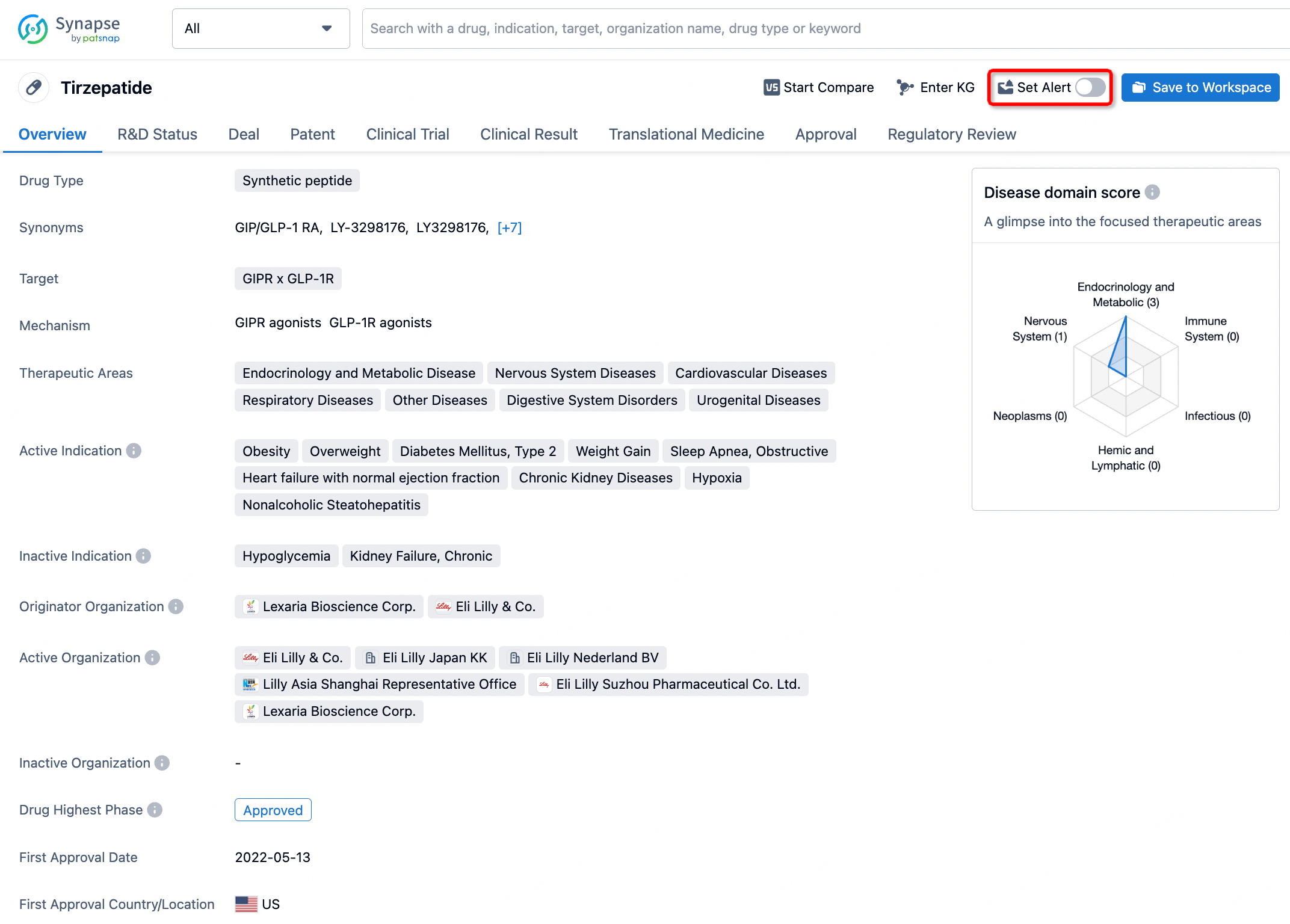
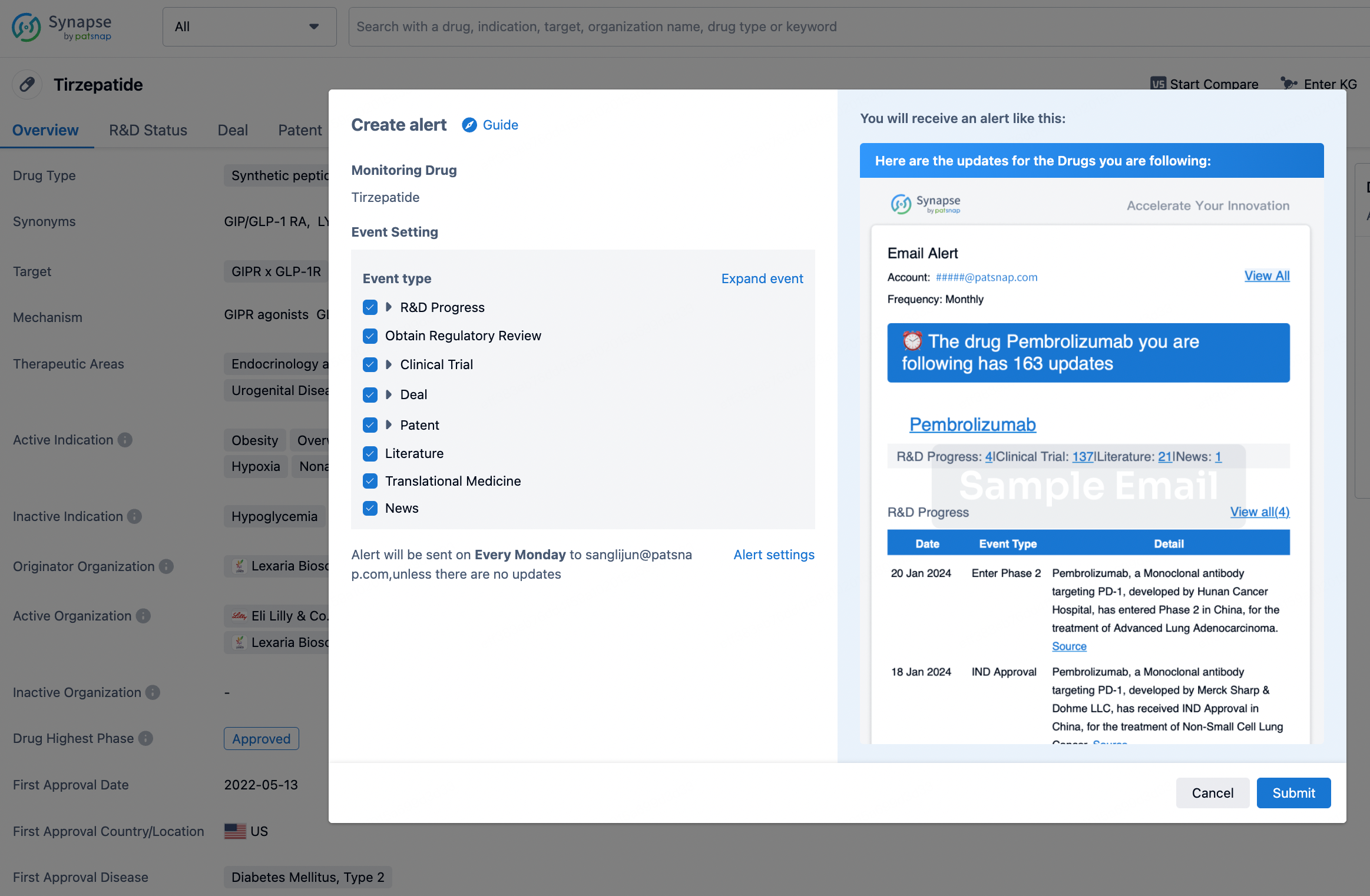
How to obtain the latest development progress of all drugs?
In the Synapse database, you can stay updated on the latest research and development advances of all drugs. This service is accessible anytime and anywhere, with updates available daily or weekly. Use the "Set Alert" function to stay informed. Click on the image below to embark on a brand new journey of drug discovery!
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.