Request Demo
What is the mechanism of Agalsidase alfa?
17 July 2024
Agalsidase alfa is an enzyme replacement therapy used primarily in the treatment of Fabry disease, a rare genetic disorder resulting from the deficiency of the enzyme alpha-galactosidase A. This deficiency leads to the accumulation of a specific type of fat, called globotriaosylceramide (Gb3 or GL-3), within various tissues and organs. Over time, this can cause a range of severe symptoms and complications, affecting the kidneys, heart, and nervous system.
The mechanism of Agalsidase alfa revolves around its role as a synthetic form of the natural enzyme alpha-galactosidase A. When administered to patients, Agalsidase alfa works to compensate for the deficient or malfunctioning endogenous enzyme. Here’s a more detailed look at the process:
1. **Enzyme Replacement**: The primary function of Agalsidase alfa is to replace the deficient alpha-galactosidase A enzyme in patients with Fabry disease. This therapeutic enzyme is produced using recombinant DNA technology, allowing it to mimic the action of the naturally occurring enzyme in the body.
2. **Substrate Breakdown**: In a healthy individual, alpha-galactosidase A breaks down Gb3 into simpler molecules that the body can easily process and eliminate. Due to the genetic mutation in Fabry disease, this breakdown does not occur effectively, leading to Gb3 accumulation. Agalsidase alfa is designed to catalyze the hydrolysis of Gb3, thereby reducing its buildup.
3. **Cellular Uptake**: Once administered intravenously, Agalsidase alfa circulates in the bloodstream and is taken up by cells through receptor-mediated endocytosis. Specifically, it binds to mannose-6-phosphate receptors on the cell surface, facilitating its internalization into lysosomes, the cell’s recycling centers.
4. **Lysosomal Function Restoration**: Inside the lysosomes, Agalsidase alfa restores the normal breakdown process of Gb3, leading to a decrease in its accumulation. By doing so, it helps to alleviate the cellular dysfunctions caused by Gb3 buildup, thereby reducing the pathological effects associated with Fabry disease.
5. **Symptom Relief and Organ Protection**: By reducing the levels of Gb3 in tissues and organs, Agalsidase alfa helps mitigate the symptoms of Fabry disease. Patients may experience relief from pain, improved kidney function, and reduced risk of heart complications. The overall quality of life can be significantly improved with regular treatment.
It is important to note that while Agalsidase alfa addresses the enzyme deficiency, it is not a cure for Fabry disease. Lifelong therapy is typically required to maintain adequate enzyme levels and manage symptoms. Additionally, the efficacy of the treatment can vary among patients, and some may experience infusion-related reactions or other side effects.
In conclusion, Agalsidase alfa serves as a critical treatment option for individuals with Fabry disease by acting as a substitute for the deficient alpha-galactosidase A enzyme. Through the breakdown of Gb3, Agalsidase alfa helps to reduce the pathological consequences of the disease, ultimately improving patient outcomes and quality of life.
The mechanism of Agalsidase alfa revolves around its role as a synthetic form of the natural enzyme alpha-galactosidase A. When administered to patients, Agalsidase alfa works to compensate for the deficient or malfunctioning endogenous enzyme. Here’s a more detailed look at the process:
1. **Enzyme Replacement**: The primary function of Agalsidase alfa is to replace the deficient alpha-galactosidase A enzyme in patients with Fabry disease. This therapeutic enzyme is produced using recombinant DNA technology, allowing it to mimic the action of the naturally occurring enzyme in the body.
2. **Substrate Breakdown**: In a healthy individual, alpha-galactosidase A breaks down Gb3 into simpler molecules that the body can easily process and eliminate. Due to the genetic mutation in Fabry disease, this breakdown does not occur effectively, leading to Gb3 accumulation. Agalsidase alfa is designed to catalyze the hydrolysis of Gb3, thereby reducing its buildup.
3. **Cellular Uptake**: Once administered intravenously, Agalsidase alfa circulates in the bloodstream and is taken up by cells through receptor-mediated endocytosis. Specifically, it binds to mannose-6-phosphate receptors on the cell surface, facilitating its internalization into lysosomes, the cell’s recycling centers.
4. **Lysosomal Function Restoration**: Inside the lysosomes, Agalsidase alfa restores the normal breakdown process of Gb3, leading to a decrease in its accumulation. By doing so, it helps to alleviate the cellular dysfunctions caused by Gb3 buildup, thereby reducing the pathological effects associated with Fabry disease.
5. **Symptom Relief and Organ Protection**: By reducing the levels of Gb3 in tissues and organs, Agalsidase alfa helps mitigate the symptoms of Fabry disease. Patients may experience relief from pain, improved kidney function, and reduced risk of heart complications. The overall quality of life can be significantly improved with regular treatment.
It is important to note that while Agalsidase alfa addresses the enzyme deficiency, it is not a cure for Fabry disease. Lifelong therapy is typically required to maintain adequate enzyme levels and manage symptoms. Additionally, the efficacy of the treatment can vary among patients, and some may experience infusion-related reactions or other side effects.
In conclusion, Agalsidase alfa serves as a critical treatment option for individuals with Fabry disease by acting as a substitute for the deficient alpha-galactosidase A enzyme. Through the breakdown of Gb3, Agalsidase alfa helps to reduce the pathological consequences of the disease, ultimately improving patient outcomes and quality of life.
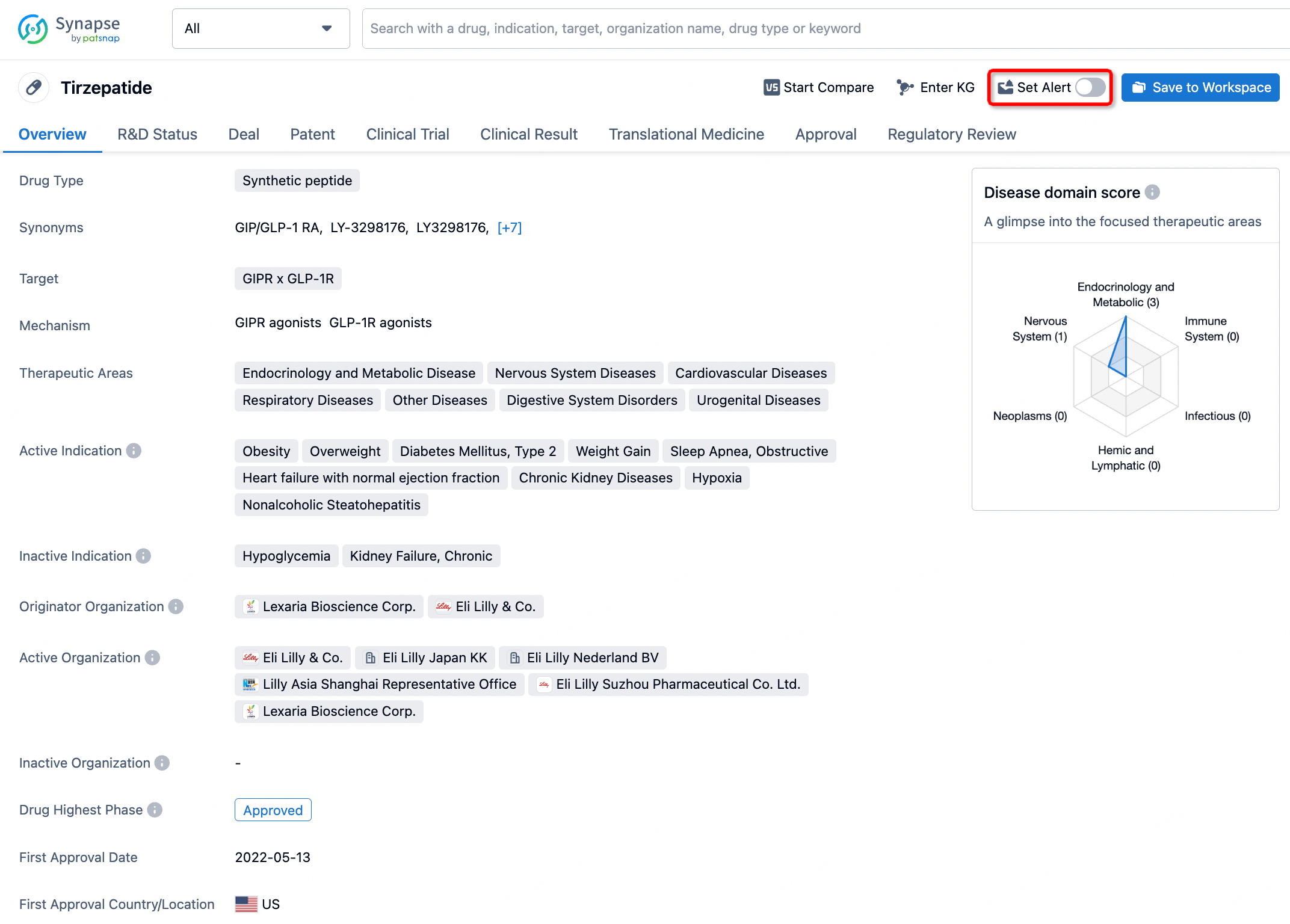
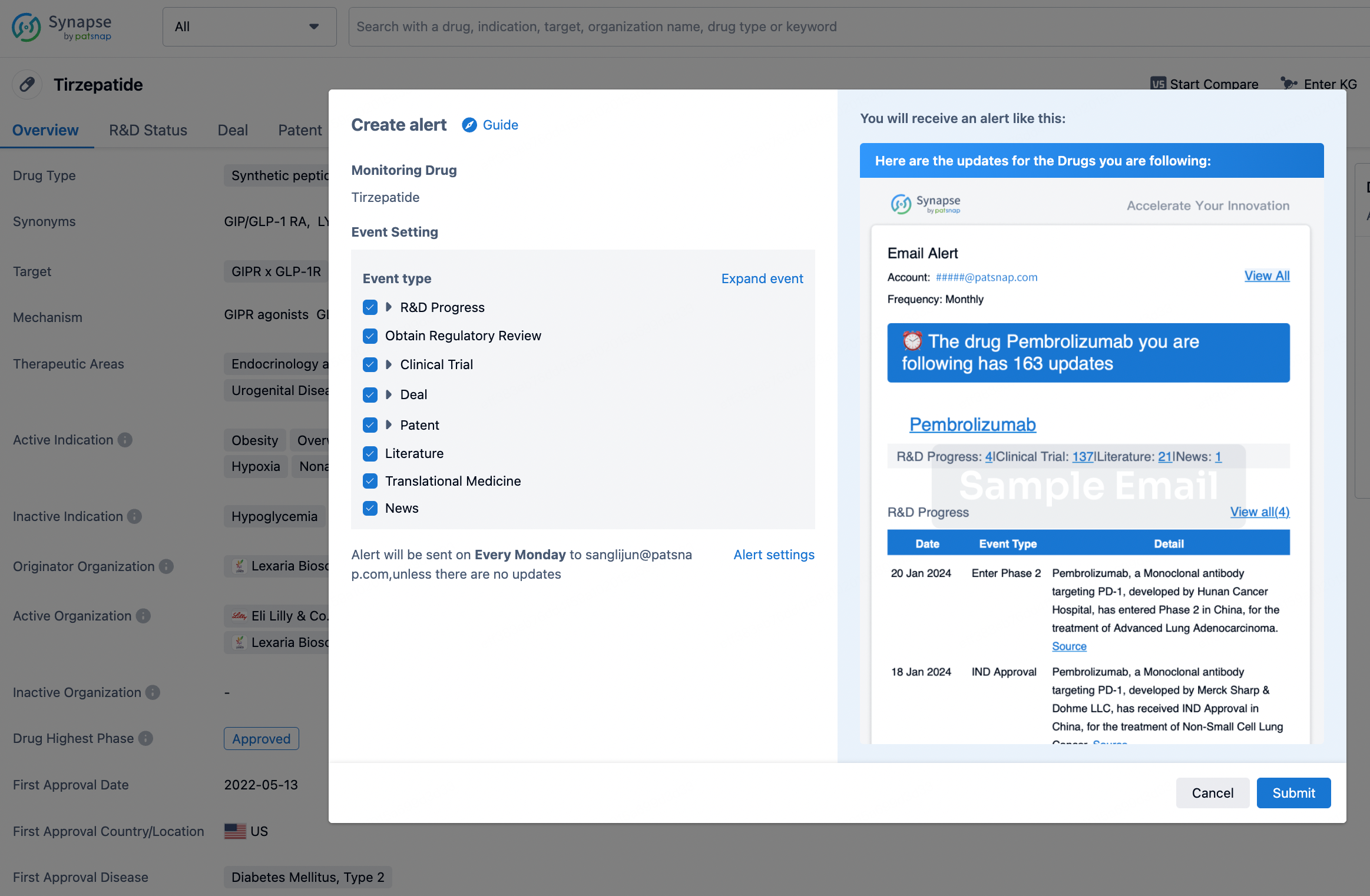
How to obtain the latest development progress of all drugs?
In the Synapse database, you can stay updated on the latest research and development advances of all drugs. This service is accessible anytime and anywhere, with updates available daily or weekly. Use the "Set Alert" function to stay informed. Click on the image below to embark on a brand new journey of drug discovery!
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.