Request Demo
What is the mechanism of Alglucosidase Alfa?
17 July 2024
Alglucosidase alfa is an enzyme replacement therapy (ERT) primarily used to treat Pompe disease, a rare genetic disorder. This condition is characterized by a deficiency of the enzyme acid alpha-glucosidase (GAA), which is responsible for breaking down glycogen into glucose within lysosomes. The absence or malfunction of GAA leads to the accumulation of glycogen in cells, particularly affecting muscle and heart tissues. Understanding the mechanism of action of alglucosidase alfa is crucial for comprehending how it alleviates the symptoms of Pompe disease.
Alglucosidase alfa is a recombinant form of the human enzyme acid alpha-glucosidase. It is produced through recombinant DNA technology using Chinese hamster ovary (CHO) cells. The primary mechanism of alglucosidase alfa involves replacing the deficient or absent GAA enzyme in patients with Pompe disease. Here is a detailed explanation of its mechanism:
1. **Enzyme Replacement**: Alglucosidase alfa is administered via intravenous infusion. Once infused into the bloodstream, it circulates throughout the body and is taken up by cells via the mannose-6-phosphate (M6P) receptor-mediated endocytosis pathway. Lysosomes, which are cellular organelles containing enzymes responsible for breaking down various biomolecules, take up the enzyme.
2. **Glycogen Breakdown**: Inside the lysosomes, alglucosidase alfa catalyzes the hydrolysis of glycogen into glucose. This is the same reaction that the naturally occurring GAA enzyme would perform. By breaking down glycogen, alglucosidase alfa helps to reduce the pathological accumulation of glycogen within the lysosomes of muscle and heart cells.
3. **Improvement of Cellular Function**: The breakdown of accumulated glycogen alleviates the cellular and tissue damage caused by its buildup. This results in improved cellular function and overall tissue health. For patients, this translates into improved muscle strength and respiratory function, as well as a reduced risk of heart complications, which are common in Pompe disease.
4. **Continuous Treatment Requirement**: Given that Pompe disease is a chronic and lifelong condition, enzyme replacement therapy with alglucosidase alfa is not a cure but a treatment. Patients typically require regular infusions, often bi-weekly, to maintain enzyme levels sufficient to manage glycogen accumulation and its associated symptoms.
5. **Long-term Outcomes**: Clinical studies have shown that alglucosidase alfa can significantly improve the quality of life for Pompe disease patients. It can enhance motor function, increase survival rates in infantile-onset Pompe disease, and stabilize or improve respiratory function in late-onset Pompe disease. However, the extent of benefit can vary based on factors such as the type of Pompe disease, age at the start of treatment, and the severity of the condition.
In summary, alglucosidase alfa works by replacing the deficient GAA enzyme in patients with Pompe disease, facilitating the breakdown of glycogen within lysosomes. This mechanism helps to mitigate the symptoms and complications associated with glycogen buildup, offering significant therapeutic benefits to those affected by this rare genetic disorder. Regular treatment with alglucosidase alfa is essential for managing the chronic nature of Pompe disease and improving patients' quality of life.
Alglucosidase alfa is a recombinant form of the human enzyme acid alpha-glucosidase. It is produced through recombinant DNA technology using Chinese hamster ovary (CHO) cells. The primary mechanism of alglucosidase alfa involves replacing the deficient or absent GAA enzyme in patients with Pompe disease. Here is a detailed explanation of its mechanism:
1. **Enzyme Replacement**: Alglucosidase alfa is administered via intravenous infusion. Once infused into the bloodstream, it circulates throughout the body and is taken up by cells via the mannose-6-phosphate (M6P) receptor-mediated endocytosis pathway. Lysosomes, which are cellular organelles containing enzymes responsible for breaking down various biomolecules, take up the enzyme.
2. **Glycogen Breakdown**: Inside the lysosomes, alglucosidase alfa catalyzes the hydrolysis of glycogen into glucose. This is the same reaction that the naturally occurring GAA enzyme would perform. By breaking down glycogen, alglucosidase alfa helps to reduce the pathological accumulation of glycogen within the lysosomes of muscle and heart cells.
3. **Improvement of Cellular Function**: The breakdown of accumulated glycogen alleviates the cellular and tissue damage caused by its buildup. This results in improved cellular function and overall tissue health. For patients, this translates into improved muscle strength and respiratory function, as well as a reduced risk of heart complications, which are common in Pompe disease.
4. **Continuous Treatment Requirement**: Given that Pompe disease is a chronic and lifelong condition, enzyme replacement therapy with alglucosidase alfa is not a cure but a treatment. Patients typically require regular infusions, often bi-weekly, to maintain enzyme levels sufficient to manage glycogen accumulation and its associated symptoms.
5. **Long-term Outcomes**: Clinical studies have shown that alglucosidase alfa can significantly improve the quality of life for Pompe disease patients. It can enhance motor function, increase survival rates in infantile-onset Pompe disease, and stabilize or improve respiratory function in late-onset Pompe disease. However, the extent of benefit can vary based on factors such as the type of Pompe disease, age at the start of treatment, and the severity of the condition.
In summary, alglucosidase alfa works by replacing the deficient GAA enzyme in patients with Pompe disease, facilitating the breakdown of glycogen within lysosomes. This mechanism helps to mitigate the symptoms and complications associated with glycogen buildup, offering significant therapeutic benefits to those affected by this rare genetic disorder. Regular treatment with alglucosidase alfa is essential for managing the chronic nature of Pompe disease and improving patients' quality of life.
How to obtain the latest development progress of all drugs?
In the Synapse database, you can stay updated on the latest research and development advances of all drugs. This service is accessible anytime and anywhere, with updates available daily or weekly. Use the "Set Alert" function to stay informed. Click on the image below to embark on a brand new journey of drug discovery!
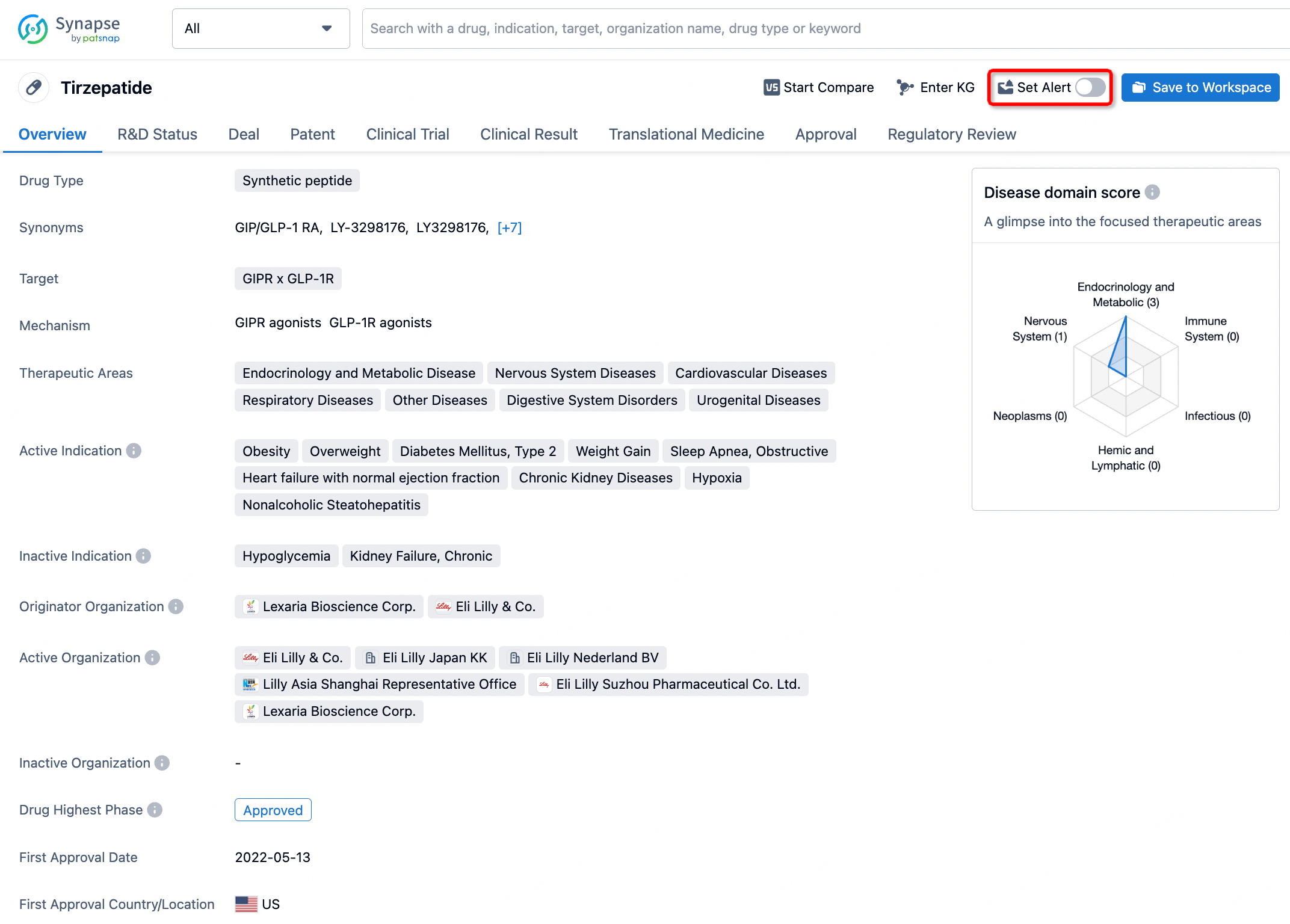
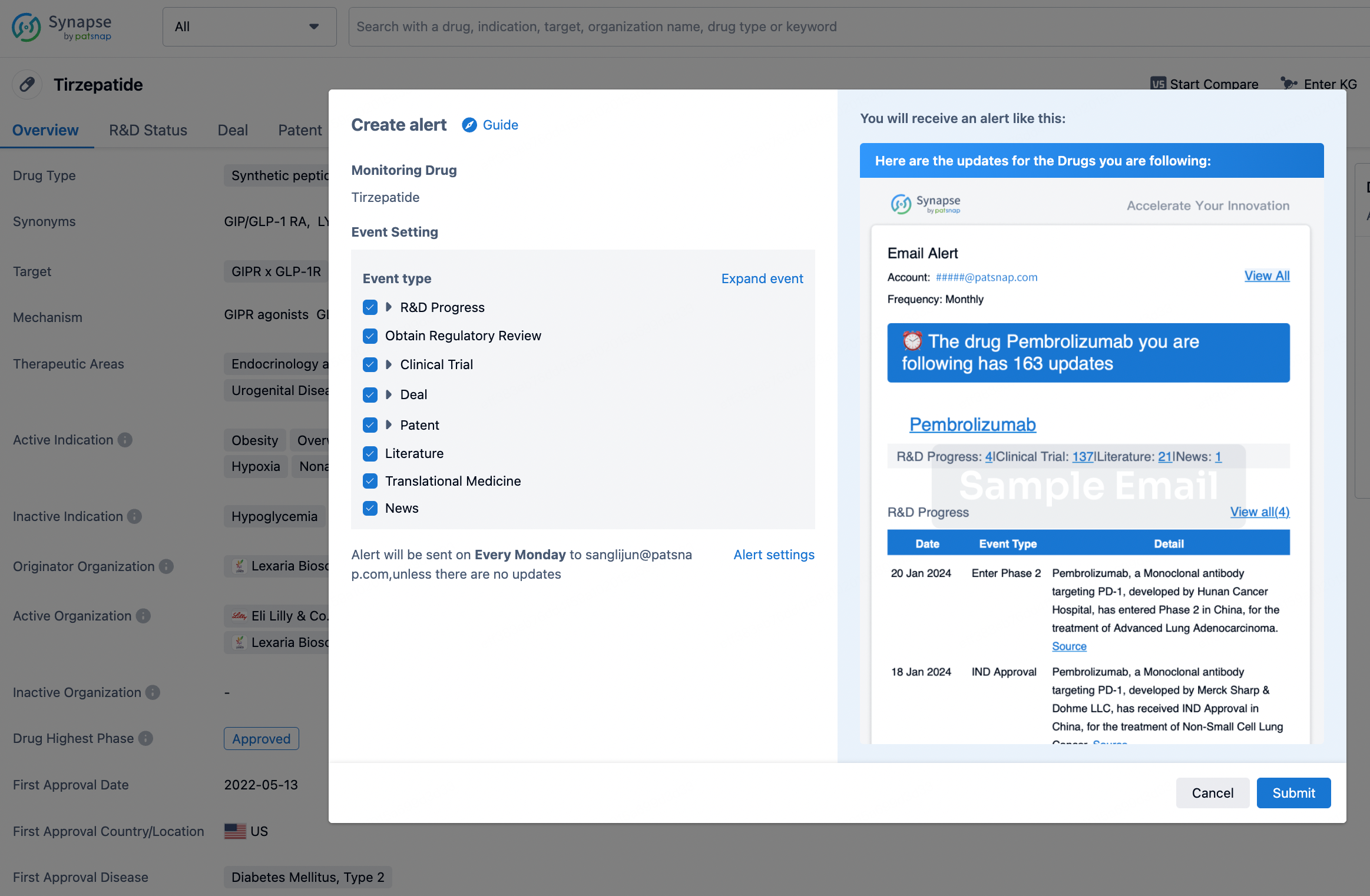
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.