Request Demo
What is the mechanism of Ataluren?
17 July 2024
Ataluren, also known as Translarna, is a pioneering pharmacological treatment specifically designed for genetic disorders caused by nonsense mutations. It’s particularly noteworthy for its application in treating Duchenne muscular dystrophy (DMD), a severe type of muscular dystrophy characterized by rapid progression of muscle degeneration. Understanding the mechanism of Ataluren requires delving into the genetic basis of the diseases it targets and how the drug interacts at the molecular level.
Genetic disorders often arise from mutations in the DNA sequence. Nonsense mutations, a specific type of mutation, introduce a premature stop codon within the coding sequence of a gene. Normally, stop codons signal the end of protein synthesis during translation, a process where ribosomes synthesize proteins based on the mRNA sequence. A premature stop codon results in truncated, non-functional, or partially functional proteins, which can lead to severe cellular dysfunction and disease.
Ataluren operates by promoting read-through of these premature stop codons during translation. Essentially, it allows the ribosome to bypass the erroneous stop signal and continue synthesizing the full-length protein. The drug selectively induces read-through only at premature stop codons without affecting normal stop codons, thus ensuring the production of functional proteins without causing harmful elongation of normal proteins.
The exact molecular interaction between Ataluren and the translational machinery is intricate. Ataluren is believed to bind to the ribosomal RNA or specific translation factors, subtly altering their conformation to reduce the efficiency of recognizing premature stop codons. This alteration enables tRNA molecules to incorporate amino acids at the position of the nonsense mutation, thereby permitting the continuation of the polypeptide chain assembly.
Clinical trials have demonstrated that Ataluren can restore sufficient levels of functional dystrophin protein in patients with nonsense mutation-mediated DMD. Dystrophin is crucial for maintaining the integrity and function of muscle cells, and its absence leads to the muscle degradation observed in DMD. By facilitating the production of functional dystrophin, Ataluren helps to stabilize muscle cell membranes, reduce inflammation, and slow disease progression.
Ataluren has shown promise beyond DMD, with potential applications in other genetic disorders caused by nonsense mutations, such as certain forms of cystic fibrosis and specific types of hemophilia. Its broad applicability stems from the common mechanism of action—read-through of premature stop codons—which is a genetic anomaly shared across various diseases.
The safety profile of Ataluren has been a critical aspect of its clinical evaluation. It’s generally well-tolerated, with most adverse effects being mild to moderate, such as gastrointestinal disturbances and headaches. The specificity of Ataluren’s action minimizes risks associated with widespread disruptions in normal protein synthesis, a significant consideration in its therapeutic use.
In conclusion, Ataluren represents a significant advancement in the treatment of genetic disorders caused by nonsense mutations. Its ability to promote the read-through of premature stop codons and enable the production of functional proteins addresses the root cause of these diseases at the molecular level. As research continues, Ataluren’s mechanism provides a valuable framework for developing future treatments for a broader array of genetic conditions, underscoring the importance of targeted molecular therapies in modern medicine.
Genetic disorders often arise from mutations in the DNA sequence. Nonsense mutations, a specific type of mutation, introduce a premature stop codon within the coding sequence of a gene. Normally, stop codons signal the end of protein synthesis during translation, a process where ribosomes synthesize proteins based on the mRNA sequence. A premature stop codon results in truncated, non-functional, or partially functional proteins, which can lead to severe cellular dysfunction and disease.
Ataluren operates by promoting read-through of these premature stop codons during translation. Essentially, it allows the ribosome to bypass the erroneous stop signal and continue synthesizing the full-length protein. The drug selectively induces read-through only at premature stop codons without affecting normal stop codons, thus ensuring the production of functional proteins without causing harmful elongation of normal proteins.
The exact molecular interaction between Ataluren and the translational machinery is intricate. Ataluren is believed to bind to the ribosomal RNA or specific translation factors, subtly altering their conformation to reduce the efficiency of recognizing premature stop codons. This alteration enables tRNA molecules to incorporate amino acids at the position of the nonsense mutation, thereby permitting the continuation of the polypeptide chain assembly.
Clinical trials have demonstrated that Ataluren can restore sufficient levels of functional dystrophin protein in patients with nonsense mutation-mediated DMD. Dystrophin is crucial for maintaining the integrity and function of muscle cells, and its absence leads to the muscle degradation observed in DMD. By facilitating the production of functional dystrophin, Ataluren helps to stabilize muscle cell membranes, reduce inflammation, and slow disease progression.
Ataluren has shown promise beyond DMD, with potential applications in other genetic disorders caused by nonsense mutations, such as certain forms of cystic fibrosis and specific types of hemophilia. Its broad applicability stems from the common mechanism of action—read-through of premature stop codons—which is a genetic anomaly shared across various diseases.
The safety profile of Ataluren has been a critical aspect of its clinical evaluation. It’s generally well-tolerated, with most adverse effects being mild to moderate, such as gastrointestinal disturbances and headaches. The specificity of Ataluren’s action minimizes risks associated with widespread disruptions in normal protein synthesis, a significant consideration in its therapeutic use.
In conclusion, Ataluren represents a significant advancement in the treatment of genetic disorders caused by nonsense mutations. Its ability to promote the read-through of premature stop codons and enable the production of functional proteins addresses the root cause of these diseases at the molecular level. As research continues, Ataluren’s mechanism provides a valuable framework for developing future treatments for a broader array of genetic conditions, underscoring the importance of targeted molecular therapies in modern medicine.
How to obtain the latest development progress of all drugs?
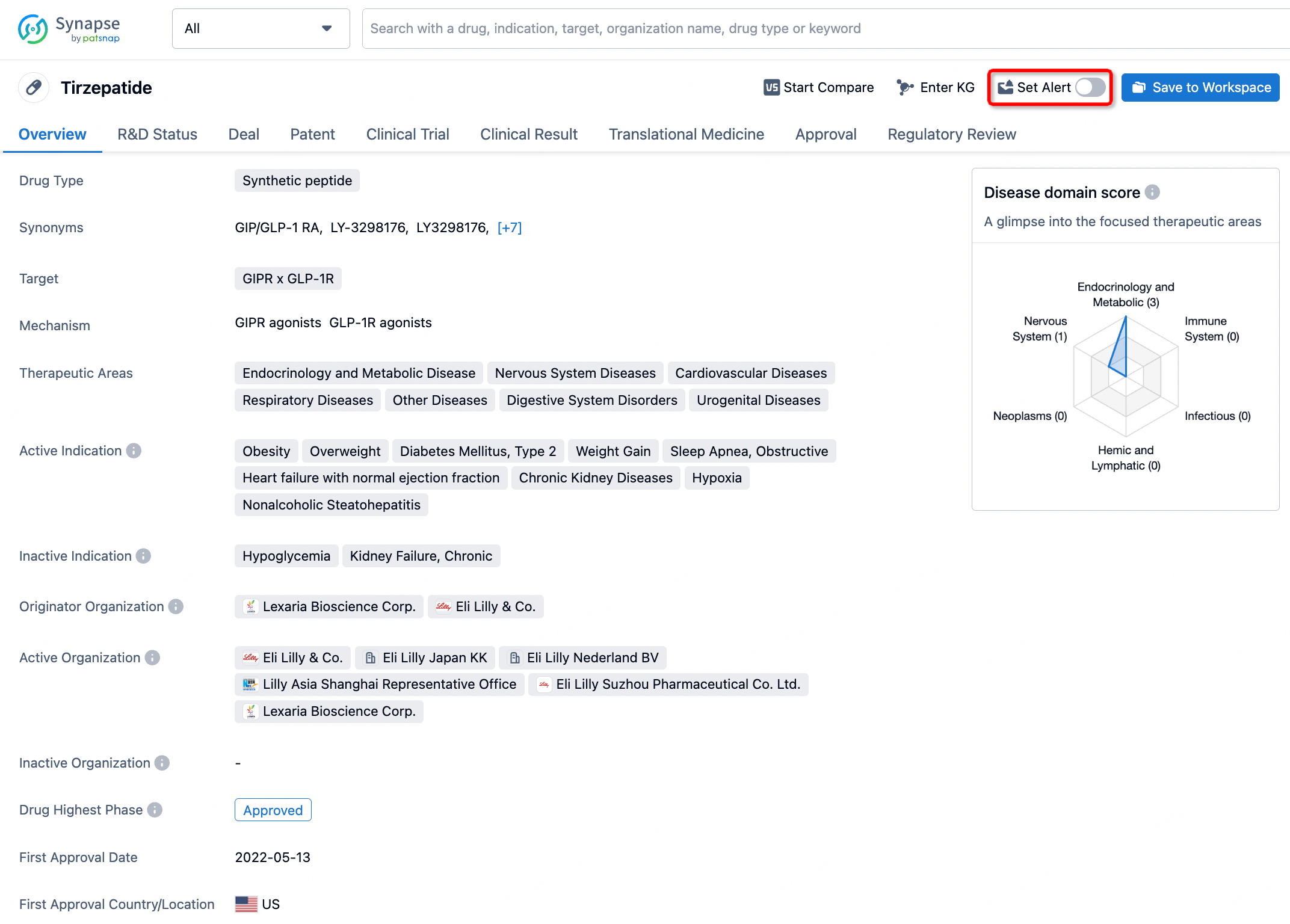
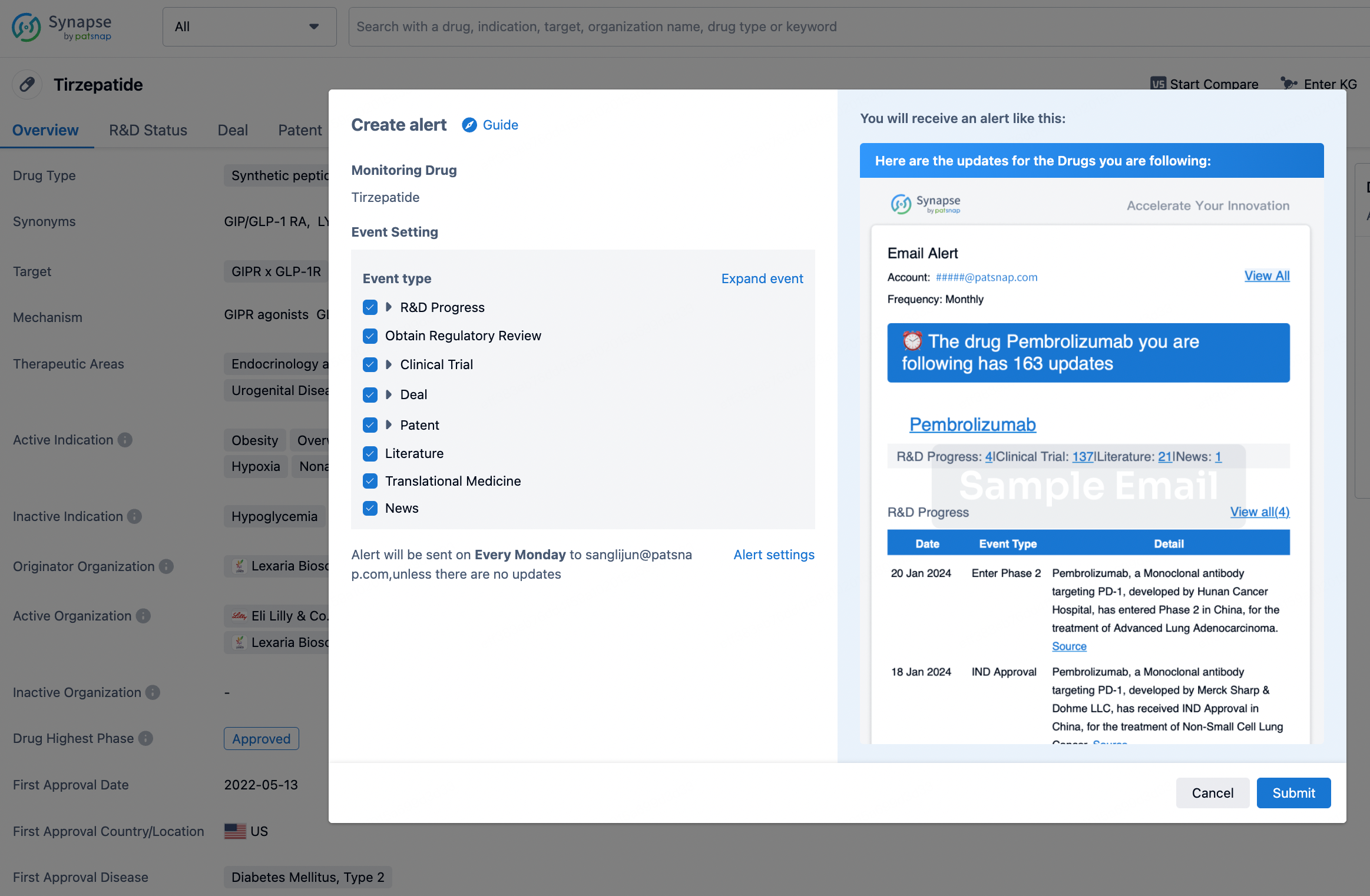
In the Synapse database, you can stay updated on the latest research and development advances of all drugs. This service is accessible anytime and anywhere, with updates available daily or weekly. Use the "Set Alert" function to stay informed. Click on the image below to embark on a brand new journey of drug discovery!
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.