Request Demo
What is the mechanism of Avalglucosidase alfa?
17 July 2024
Avalglucosidase alfa is a cutting-edge therapeutic agent designed to address a specific genetic disorder known as Pompe disease. Understanding its mechanism of action is crucial to appreciate how it aids in alleviating this condition.
Pompe disease is a lysosomal storage disorder caused by a deficiency of the enzyme acid alpha-glucosidase (GAA). This enzyme is responsible for breaking down glycogen, a complex sugar stored in cells, into glucose, which cells use for energy. When GAA is deficient or absent, glycogen accumulates in lysosomes, the recycling centers of cells, particularly within muscle tissues. This accumulation leads to progressive muscle weakness and respiratory difficulties, which are hallmarks of Pompe disease.
Avalglucosidase alfa works as an enzyme replacement therapy (ERT). Essentially, it supplements the deficient or missing GAA enzyme in patients with Pompe disease. The drug is a recombinant form of human GAA, meaning it is produced using DNA technology to create a synthetic version of the naturally occurring enzyme. Once administered, it targets and enters lysosomes, the organelles overloaded with glycogen in Pompe patients.
The mechanism by which Avalglucosidase alfa enters lysosomes is sophisticated. The enzyme contains a specific oligosaccharide (a carbohydrate chain) with mannose-6-phosphate (M6P) residues. These M6P residues are critical for the enzyme's uptake into cells. Cells have mannose-6-phosphate receptors (M6PR) on their surface, which recognize and bind to the M6P-tagged enzyme. Once bound, the enzyme-receptor complex is internalized into the cell and transported to the lysosome.
Inside the lysosome, Avalglucosidase alfa catalyzes the hydrolysis of glycogen into glucose. This process reduces the glycogen buildup within lysosomes, thereby decreasing the detrimental effects of its accumulation. By restoring the normal breakdown of glycogen, the therapy helps improve muscle function and respiratory capabilities, addressing the core symptoms of Pompe disease.
To maximize its effectiveness, Avalglucosidase alfa is often administered intravenously. This method ensures that the enzyme circulates throughout the body and reaches muscle tissues where it is most needed. The frequency and dosage of the treatment depend on various factors, including the patient's weight and the severity of the disease.
In summary, Avalglucosidase alfa acts as a substitute for the deficient GAA enzyme in Pompe disease. Its action hinges on its ability to enter cells via M6P receptors, deliver its enzymatic activity within lysosomes, and facilitate the breakdown of glycogen. By doing so, it significantly mitigates the symptoms of Pompe disease, offering patients improved quality of life and better overall health outcomes.
Pompe disease is a lysosomal storage disorder caused by a deficiency of the enzyme acid alpha-glucosidase (GAA). This enzyme is responsible for breaking down glycogen, a complex sugar stored in cells, into glucose, which cells use for energy. When GAA is deficient or absent, glycogen accumulates in lysosomes, the recycling centers of cells, particularly within muscle tissues. This accumulation leads to progressive muscle weakness and respiratory difficulties, which are hallmarks of Pompe disease.
Avalglucosidase alfa works as an enzyme replacement therapy (ERT). Essentially, it supplements the deficient or missing GAA enzyme in patients with Pompe disease. The drug is a recombinant form of human GAA, meaning it is produced using DNA technology to create a synthetic version of the naturally occurring enzyme. Once administered, it targets and enters lysosomes, the organelles overloaded with glycogen in Pompe patients.
The mechanism by which Avalglucosidase alfa enters lysosomes is sophisticated. The enzyme contains a specific oligosaccharide (a carbohydrate chain) with mannose-6-phosphate (M6P) residues. These M6P residues are critical for the enzyme's uptake into cells. Cells have mannose-6-phosphate receptors (M6PR) on their surface, which recognize and bind to the M6P-tagged enzyme. Once bound, the enzyme-receptor complex is internalized into the cell and transported to the lysosome.
Inside the lysosome, Avalglucosidase alfa catalyzes the hydrolysis of glycogen into glucose. This process reduces the glycogen buildup within lysosomes, thereby decreasing the detrimental effects of its accumulation. By restoring the normal breakdown of glycogen, the therapy helps improve muscle function and respiratory capabilities, addressing the core symptoms of Pompe disease.
To maximize its effectiveness, Avalglucosidase alfa is often administered intravenously. This method ensures that the enzyme circulates throughout the body and reaches muscle tissues where it is most needed. The frequency and dosage of the treatment depend on various factors, including the patient's weight and the severity of the disease.
In summary, Avalglucosidase alfa acts as a substitute for the deficient GAA enzyme in Pompe disease. Its action hinges on its ability to enter cells via M6P receptors, deliver its enzymatic activity within lysosomes, and facilitate the breakdown of glycogen. By doing so, it significantly mitigates the symptoms of Pompe disease, offering patients improved quality of life and better overall health outcomes.
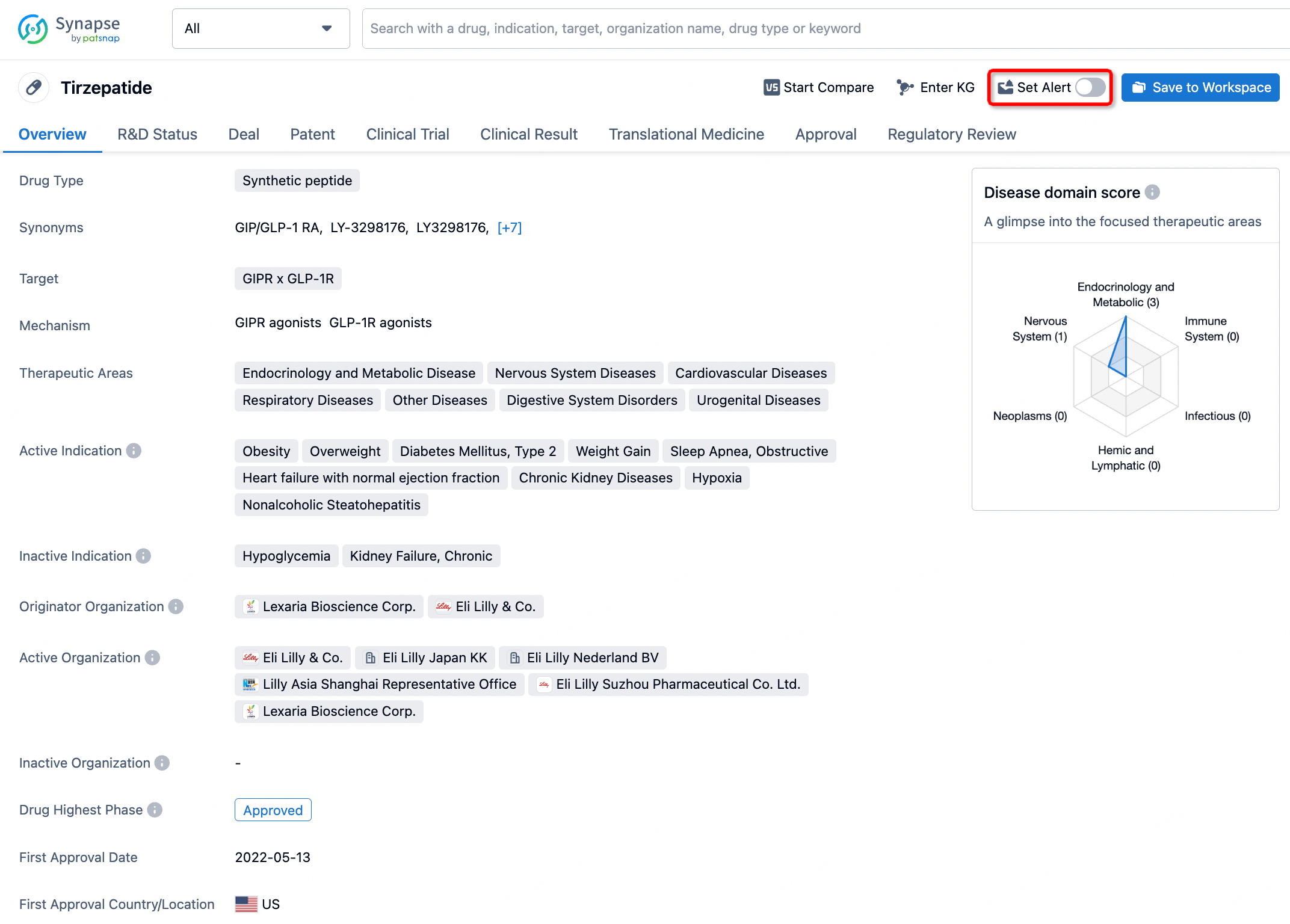
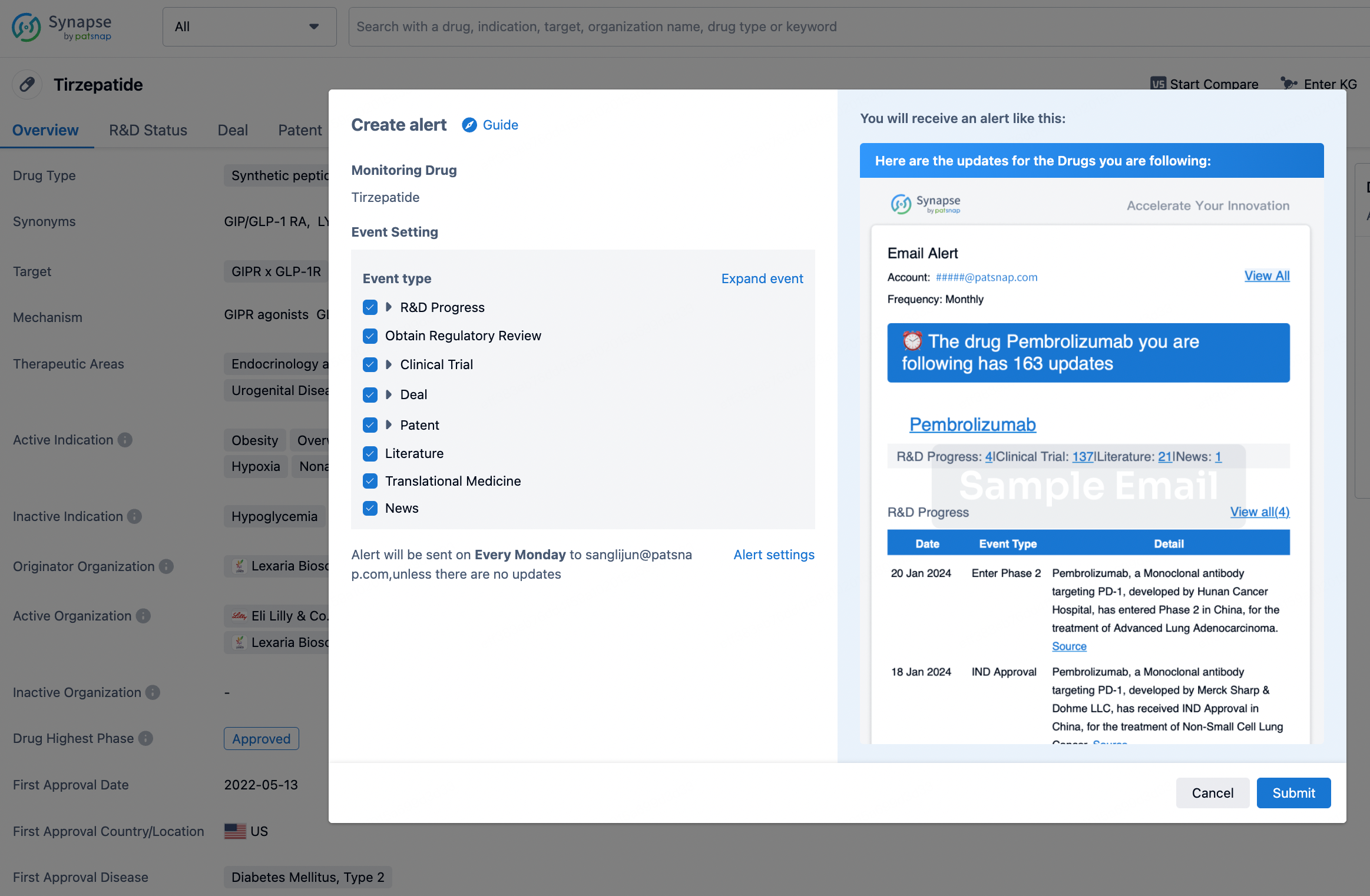
How to obtain the latest development progress of all drugs?
In the Synapse database, you can stay updated on the latest research and development advances of all drugs. This service is accessible anytime and anywhere, with updates available daily or weekly. Use the "Set Alert" function to stay informed. Click on the image below to embark on a brand new journey of drug discovery!
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.