Request Demo
What is the mechanism of Gefitinib?
17 July 2024
Gefitinib is a tyrosine kinase inhibitor (TKI) specifically targeting the epidermal growth factor receptor (EGFR) pathway, which plays a significant role in the proliferation and survival of various cancer cells. Understanding the mechanism of Gefitinib requires a closer look at its interaction with EGFR and the downstream signaling pathways that are affected.
EGFR is a transmembrane protein that, when activated by binding to its natural ligands such as epidermal growth factor (EGF) and transforming growth factor-alpha (TGF-α), undergoes a conformational change that leads to its dimerization. This dimerization activates its intrinsic tyrosine kinase activity, resulting in the autophosphorylation of tyrosine residues in the cytoplasmic domain of the receptor. These phosphorylated tyrosine residues serve as docking sites for various signaling proteins, initiating multiple downstream signaling cascades, including the RAS-RAF-MEK-ERK pathway, the PI3K-AKT pathway, and the JAK-STAT pathway, all of which are crucial for cell proliferation, survival, and differentiation.
Gefitinib works by selectively binding to the adenosine triphosphate (ATP)-binding site of the tyrosine kinase domain of EGFR. This binding inhibits the autophosphorylation of the receptor, effectively blocking the activation of the downstream signaling pathways. By inhibiting EGFR signaling, Gefitinib can reduce the proliferation of cancer cells and induce apoptosis, particularly in tumors that are dependent on EGFR signaling for growth and survival.
Gefitinib is particularly effective in non-small cell lung cancer (NSCLC) patients who have specific activating mutations in the EGFR gene. These mutations, often found in the tyrosine kinase domain of the EGFR, lead to increased sensitivity to Gefitinib. The most common mutations associated with increased sensitivity to Gefitinib are deletions in exon 19 and the L858R point mutation in exon 21. These mutations result in a constitutively active EGFR that drives oncogenesis, and Gefitinib’s ability to inhibit this aberrant signaling makes it an effective therapeutic agent in these cases.
In addition to its role in inhibiting EGFR signaling, Gefitinib has been shown to affect other cellular processes. For instance, it can inhibit angiogenesis, the process by which new blood vessels are formed, which is critical for tumor growth and metastasis. By reducing the expression of vascular endothelial growth factor (VEGF) and other pro-angiogenic factors, Gefitinib can limit the blood supply to the tumor, further inhibiting its growth.
Despite its efficacy, resistance to Gefitinib can develop, often through secondary mutations in the EGFR gene, such as the T790M mutation, which alters the binding affinity of the receptor for Gefitinib. Additionally, activation of alternative signaling pathways, such as the MET or HER2 pathways, can also contribute to resistance. Understanding these mechanisms of resistance is crucial for developing new strategies to overcome them and improve the efficacy of EGFR-targeted therapies.
In conclusion, Gefitinib works by specifically inhibiting the tyrosine kinase activity of EGFR, blocking critical signaling pathways involved in cancer cell proliferation and survival. Its effectiveness in NSCLC patients with specific EGFR mutations highlights the importance of molecular profiling in the management of cancer. However, the development of resistance remains a significant challenge, necessitating ongoing research to develop next-generation inhibitors and combination therapies to enhance treatment outcomes.
EGFR is a transmembrane protein that, when activated by binding to its natural ligands such as epidermal growth factor (EGF) and transforming growth factor-alpha (TGF-α), undergoes a conformational change that leads to its dimerization. This dimerization activates its intrinsic tyrosine kinase activity, resulting in the autophosphorylation of tyrosine residues in the cytoplasmic domain of the receptor. These phosphorylated tyrosine residues serve as docking sites for various signaling proteins, initiating multiple downstream signaling cascades, including the RAS-RAF-MEK-ERK pathway, the PI3K-AKT pathway, and the JAK-STAT pathway, all of which are crucial for cell proliferation, survival, and differentiation.
Gefitinib works by selectively binding to the adenosine triphosphate (ATP)-binding site of the tyrosine kinase domain of EGFR. This binding inhibits the autophosphorylation of the receptor, effectively blocking the activation of the downstream signaling pathways. By inhibiting EGFR signaling, Gefitinib can reduce the proliferation of cancer cells and induce apoptosis, particularly in tumors that are dependent on EGFR signaling for growth and survival.
Gefitinib is particularly effective in non-small cell lung cancer (NSCLC) patients who have specific activating mutations in the EGFR gene. These mutations, often found in the tyrosine kinase domain of the EGFR, lead to increased sensitivity to Gefitinib. The most common mutations associated with increased sensitivity to Gefitinib are deletions in exon 19 and the L858R point mutation in exon 21. These mutations result in a constitutively active EGFR that drives oncogenesis, and Gefitinib’s ability to inhibit this aberrant signaling makes it an effective therapeutic agent in these cases.
In addition to its role in inhibiting EGFR signaling, Gefitinib has been shown to affect other cellular processes. For instance, it can inhibit angiogenesis, the process by which new blood vessels are formed, which is critical for tumor growth and metastasis. By reducing the expression of vascular endothelial growth factor (VEGF) and other pro-angiogenic factors, Gefitinib can limit the blood supply to the tumor, further inhibiting its growth.
Despite its efficacy, resistance to Gefitinib can develop, often through secondary mutations in the EGFR gene, such as the T790M mutation, which alters the binding affinity of the receptor for Gefitinib. Additionally, activation of alternative signaling pathways, such as the MET or HER2 pathways, can also contribute to resistance. Understanding these mechanisms of resistance is crucial for developing new strategies to overcome them and improve the efficacy of EGFR-targeted therapies.
In conclusion, Gefitinib works by specifically inhibiting the tyrosine kinase activity of EGFR, blocking critical signaling pathways involved in cancer cell proliferation and survival. Its effectiveness in NSCLC patients with specific EGFR mutations highlights the importance of molecular profiling in the management of cancer. However, the development of resistance remains a significant challenge, necessitating ongoing research to develop next-generation inhibitors and combination therapies to enhance treatment outcomes.
How to obtain the latest development progress of all drugs?
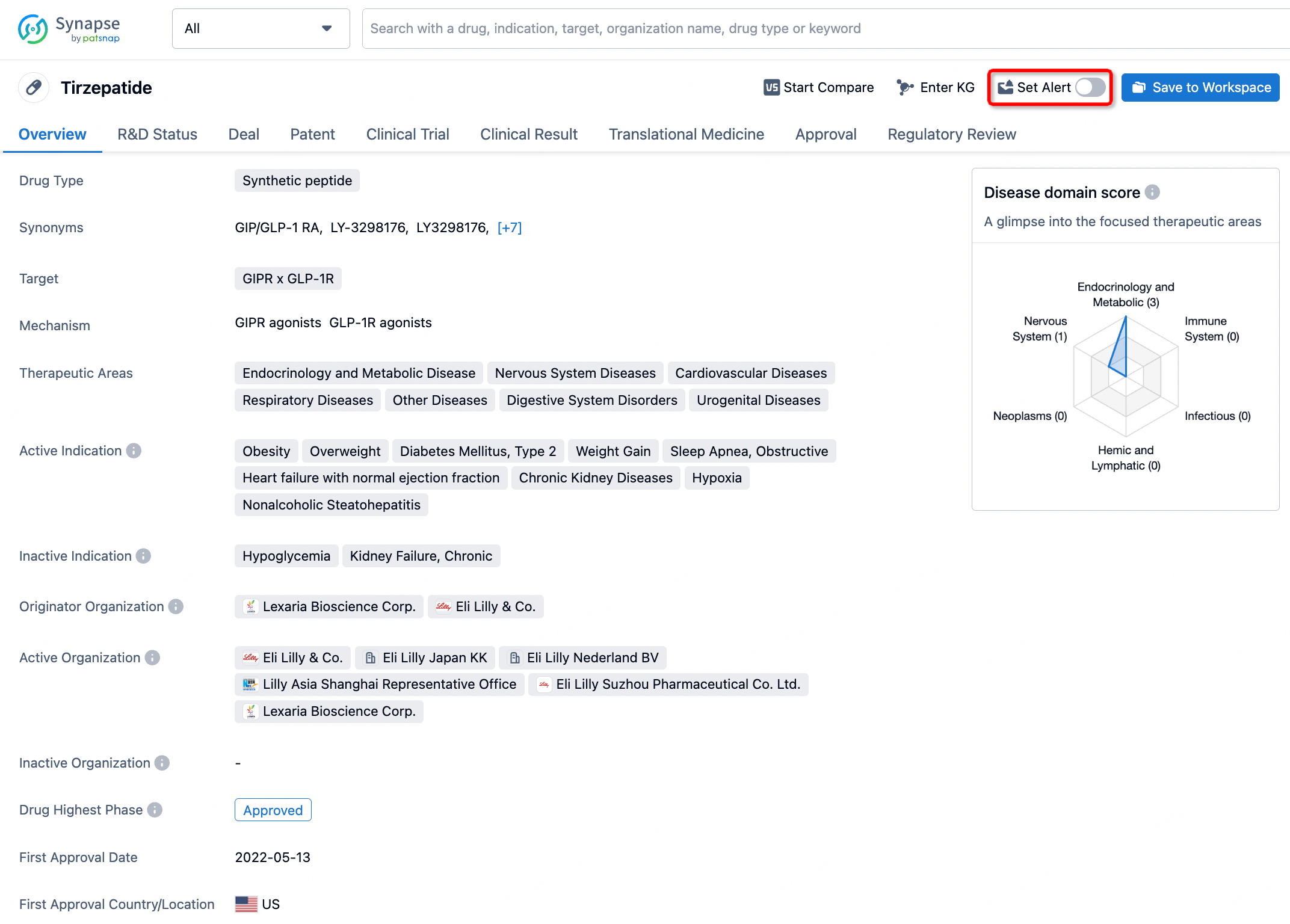
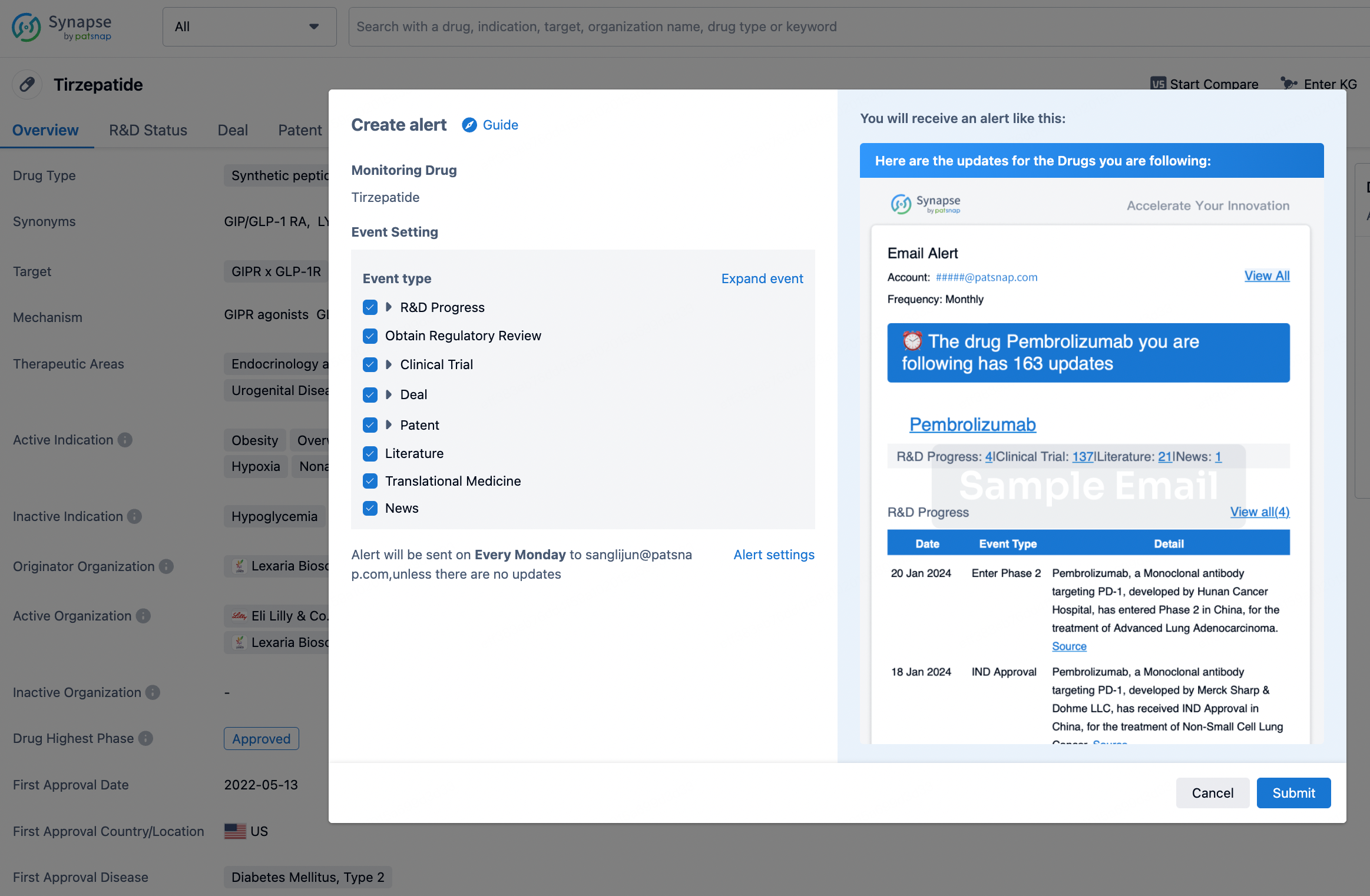
In the Synapse database, you can stay updated on the latest research and development advances of all drugs. This service is accessible anytime and anywhere, with updates available daily or weekly. Use the "Set Alert" function to stay informed. Click on the image below to embark on a brand new journey of drug discovery!
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.