Request Demo
What is the mechanism of Migalastat Hydrochloride?
17 July 2024
Migalastat Hydrochloride is a pharmacological chaperone used in the treatment of Fabry disease, a genetic disorder caused by mutations in the GLA gene resulting in deficient or dysfunctional alpha-galactosidase A (α-Gal A) enzyme. Understanding the mechanism of action of Migalastat Hydrochloride is essential for comprehending its therapeutic benefits and its role in managing Fabry disease.
Fabry disease is an X-linked lysosomal storage disorder characterized by the accumulation of globotriaosylceramide (GL-3) within lysosomes due to insufficient α-Gal A activity. The buildup of GL-3 in various tissues leads to a spectrum of symptoms including pain, renal failure, cardiac issues, and cerebrovascular complications.
Migalastat Hydrochloride operates through a mechanism known as pharmacological chaperoning. This process involves stabilizing and enhancing the function of the mutant α-Gal A enzyme. Many mutations in the GLA gene result in misfolded enzyme proteins that are typically retained in the endoplasmic reticulum (ER) and subsequently degraded rather than being transported to lysosomes where they are needed.
Migalastat selectively binds to the active site of certain mutant forms of α-Gal A, stabilizing their conformation. This binding action assists in the proper folding of the enzyme, allowing it to escape the quality control system of the ER. Consequently, the stabilized enzyme is transported to the lysosome, where it can perform its physiological function of breaking down GL-3.
The efficacy of Migalastat is mutation-specific; it is only effective in patients who have amenable mutations. These are specific mutations in the GLA gene where the resultant enzyme retains some degree of functionality and can be properly folded with the aid of Migalastat. The identification of amenable mutations is typically determined through in vitro assays that test the interaction between Migalastat and the mutant enzyme.
Once in the lysosome, the stabilized α-Gal A enzyme facilitated by Migalastat can degrade GL-3, thereby reducing its pathological accumulation. This reduction in GL-3 levels helps alleviate the cellular and tissue dysfunctions observed in Fabry disease patients, improving clinical outcomes.
In summary, Migalastat Hydrochloride acts as a pharmacological chaperone that facilitates the proper folding and lysosomal trafficking of certain mutant forms of α-Gal A enzyme in Fabry disease patients with amenable mutations. By stabilizing these mutant enzymes, Migalastat enhances their functional activity, thereby reducing the pathological accumulation of GL-3 and ameliorating disease symptoms. This targeted mechanism highlights the importance of genetic screening and personalized medicine in the effective treatment of genetic disorders like Fabry disease.
Fabry disease is an X-linked lysosomal storage disorder characterized by the accumulation of globotriaosylceramide (GL-3) within lysosomes due to insufficient α-Gal A activity. The buildup of GL-3 in various tissues leads to a spectrum of symptoms including pain, renal failure, cardiac issues, and cerebrovascular complications.
Migalastat Hydrochloride operates through a mechanism known as pharmacological chaperoning. This process involves stabilizing and enhancing the function of the mutant α-Gal A enzyme. Many mutations in the GLA gene result in misfolded enzyme proteins that are typically retained in the endoplasmic reticulum (ER) and subsequently degraded rather than being transported to lysosomes where they are needed.
Migalastat selectively binds to the active site of certain mutant forms of α-Gal A, stabilizing their conformation. This binding action assists in the proper folding of the enzyme, allowing it to escape the quality control system of the ER. Consequently, the stabilized enzyme is transported to the lysosome, where it can perform its physiological function of breaking down GL-3.
The efficacy of Migalastat is mutation-specific; it is only effective in patients who have amenable mutations. These are specific mutations in the GLA gene where the resultant enzyme retains some degree of functionality and can be properly folded with the aid of Migalastat. The identification of amenable mutations is typically determined through in vitro assays that test the interaction between Migalastat and the mutant enzyme.
Once in the lysosome, the stabilized α-Gal A enzyme facilitated by Migalastat can degrade GL-3, thereby reducing its pathological accumulation. This reduction in GL-3 levels helps alleviate the cellular and tissue dysfunctions observed in Fabry disease patients, improving clinical outcomes.
In summary, Migalastat Hydrochloride acts as a pharmacological chaperone that facilitates the proper folding and lysosomal trafficking of certain mutant forms of α-Gal A enzyme in Fabry disease patients with amenable mutations. By stabilizing these mutant enzymes, Migalastat enhances their functional activity, thereby reducing the pathological accumulation of GL-3 and ameliorating disease symptoms. This targeted mechanism highlights the importance of genetic screening and personalized medicine in the effective treatment of genetic disorders like Fabry disease.
How to obtain the latest development progress of all drugs?
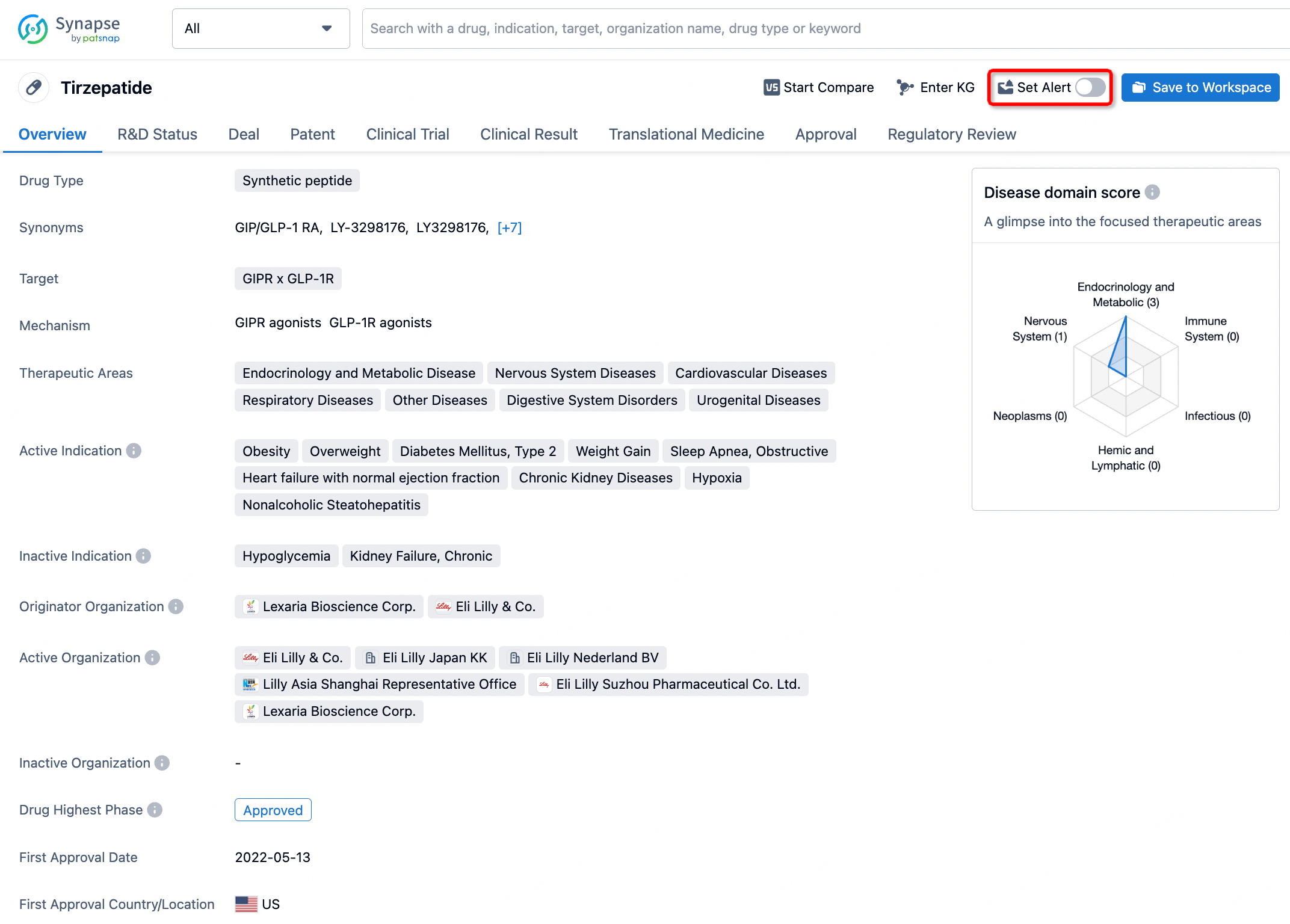
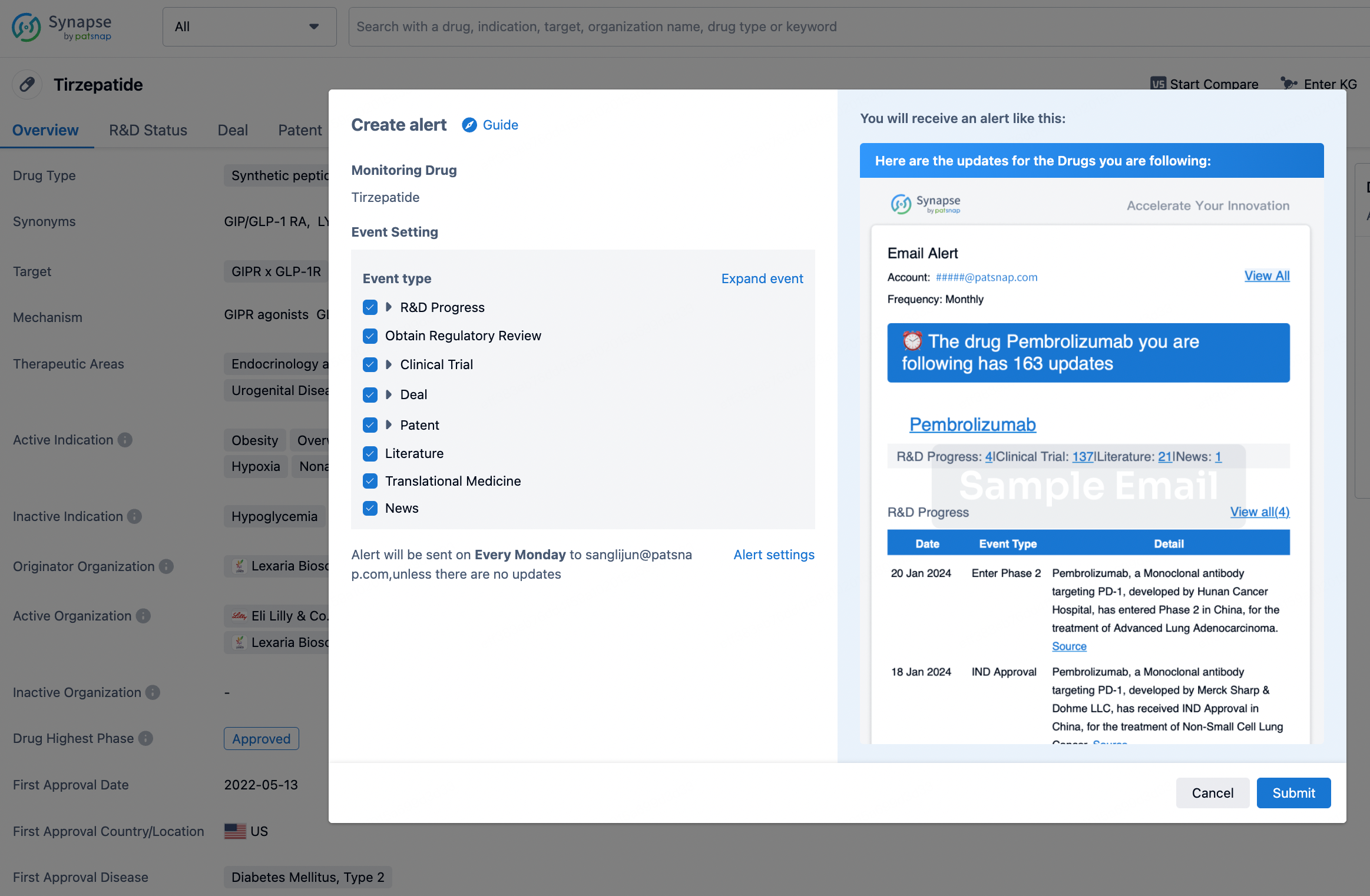
In the Synapse database, you can stay updated on the latest research and development advances of all drugs. This service is accessible anytime and anywhere, with updates available daily or weekly. Use the "Set Alert" function to stay informed. Click on the image below to embark on a brand new journey of drug discovery!
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.