Request Demo
What is the mechanism of Risdiplam?
17 July 2024
Risdiplam is a medication used to treat spinal muscular atrophy (SMA), a genetic disorder characterized by the loss of motor neurons, leading to muscle wasting and weakness. To understand the mechanism of Risdiplam, it is essential to delve into the genetic and molecular basis of SMA and how this drug intervenes in the disease process.
Spinal muscular atrophy is primarily caused by mutations in the survival motor neuron 1 (SMN1) gene. This gene produces a protein called SMN, which is critical for the maintenance and function of motor neurons. In individuals with SMA, the SMN1 gene is either deleted or mutated, leading to a significant reduction in the levels of functional SMN protein. Humans have a second gene, SMN2, which is nearly identical to SMN1; however, due to a single nucleotide difference, most of the SMN protein produced from SMN2 is truncated and non-functional. Only about 10% of the protein produced by SMN2 is full-length and functional.
Risdiplam is designed to address this problem by modifying the splicing of the SMN2 pre-mRNA, thereby increasing the production of full-length, functional SMN protein. It is an orally administered, small-molecule drug that acts as a splicing modifier. The mechanism by which Risdiplam operates involves binding to specific sites on the SMN2 pre-mRNA, influencing the splicing machinery to include exon 7, which is typically excluded in the truncated version of the SMN protein produced by SMN2.
By promoting the inclusion of exon 7, Risdiplam effectively increases the proportion of full-length SMN protein generated by the SMN2 gene. This increase in functional SMN protein helps to compensate for the deficiency caused by the mutation or deletion of the SMN1 gene, thereby improving the survival and function of motor neurons.
In clinical studies, Risdiplam has demonstrated significant efficacy in increasing SMN protein levels in patients with SMA. The resultant increase in functional SMN protein leads to improvements in motor function and delays in the progression of the disease. Importantly, Risdiplam has been shown to be effective across a range of ages and stages of SMA, making it a versatile treatment option.
Moreover, the oral administration of Risdiplam offers a convenient alternative to other treatment modalities that may require intrathecal or intravenous delivery, thereby improving patient compliance and quality of life. The broad distribution of Risdiplam in the body, including its ability to cross the blood-brain barrier, ensures that it can reach the central nervous system where motor neurons reside, as well as peripheral tissues affected by SMA.
In summary, Risdiplam works by modifying the splicing of SMN2 pre-mRNA to increase the production of full-length, functional SMN protein, thereby compensating for the deficiency caused by the mutation or deletion of the SMN1 gene. Its ability to improve motor function and delay disease progression in SMA patients underscores its efficacy and represents a significant advancement in the treatment of this debilitating genetic disorder.
Spinal muscular atrophy is primarily caused by mutations in the survival motor neuron 1 (SMN1) gene. This gene produces a protein called SMN, which is critical for the maintenance and function of motor neurons. In individuals with SMA, the SMN1 gene is either deleted or mutated, leading to a significant reduction in the levels of functional SMN protein. Humans have a second gene, SMN2, which is nearly identical to SMN1; however, due to a single nucleotide difference, most of the SMN protein produced from SMN2 is truncated and non-functional. Only about 10% of the protein produced by SMN2 is full-length and functional.
Risdiplam is designed to address this problem by modifying the splicing of the SMN2 pre-mRNA, thereby increasing the production of full-length, functional SMN protein. It is an orally administered, small-molecule drug that acts as a splicing modifier. The mechanism by which Risdiplam operates involves binding to specific sites on the SMN2 pre-mRNA, influencing the splicing machinery to include exon 7, which is typically excluded in the truncated version of the SMN protein produced by SMN2.
By promoting the inclusion of exon 7, Risdiplam effectively increases the proportion of full-length SMN protein generated by the SMN2 gene. This increase in functional SMN protein helps to compensate for the deficiency caused by the mutation or deletion of the SMN1 gene, thereby improving the survival and function of motor neurons.
In clinical studies, Risdiplam has demonstrated significant efficacy in increasing SMN protein levels in patients with SMA. The resultant increase in functional SMN protein leads to improvements in motor function and delays in the progression of the disease. Importantly, Risdiplam has been shown to be effective across a range of ages and stages of SMA, making it a versatile treatment option.
Moreover, the oral administration of Risdiplam offers a convenient alternative to other treatment modalities that may require intrathecal or intravenous delivery, thereby improving patient compliance and quality of life. The broad distribution of Risdiplam in the body, including its ability to cross the blood-brain barrier, ensures that it can reach the central nervous system where motor neurons reside, as well as peripheral tissues affected by SMA.
In summary, Risdiplam works by modifying the splicing of SMN2 pre-mRNA to increase the production of full-length, functional SMN protein, thereby compensating for the deficiency caused by the mutation or deletion of the SMN1 gene. Its ability to improve motor function and delay disease progression in SMA patients underscores its efficacy and represents a significant advancement in the treatment of this debilitating genetic disorder.
How to obtain the latest development progress of all drugs?
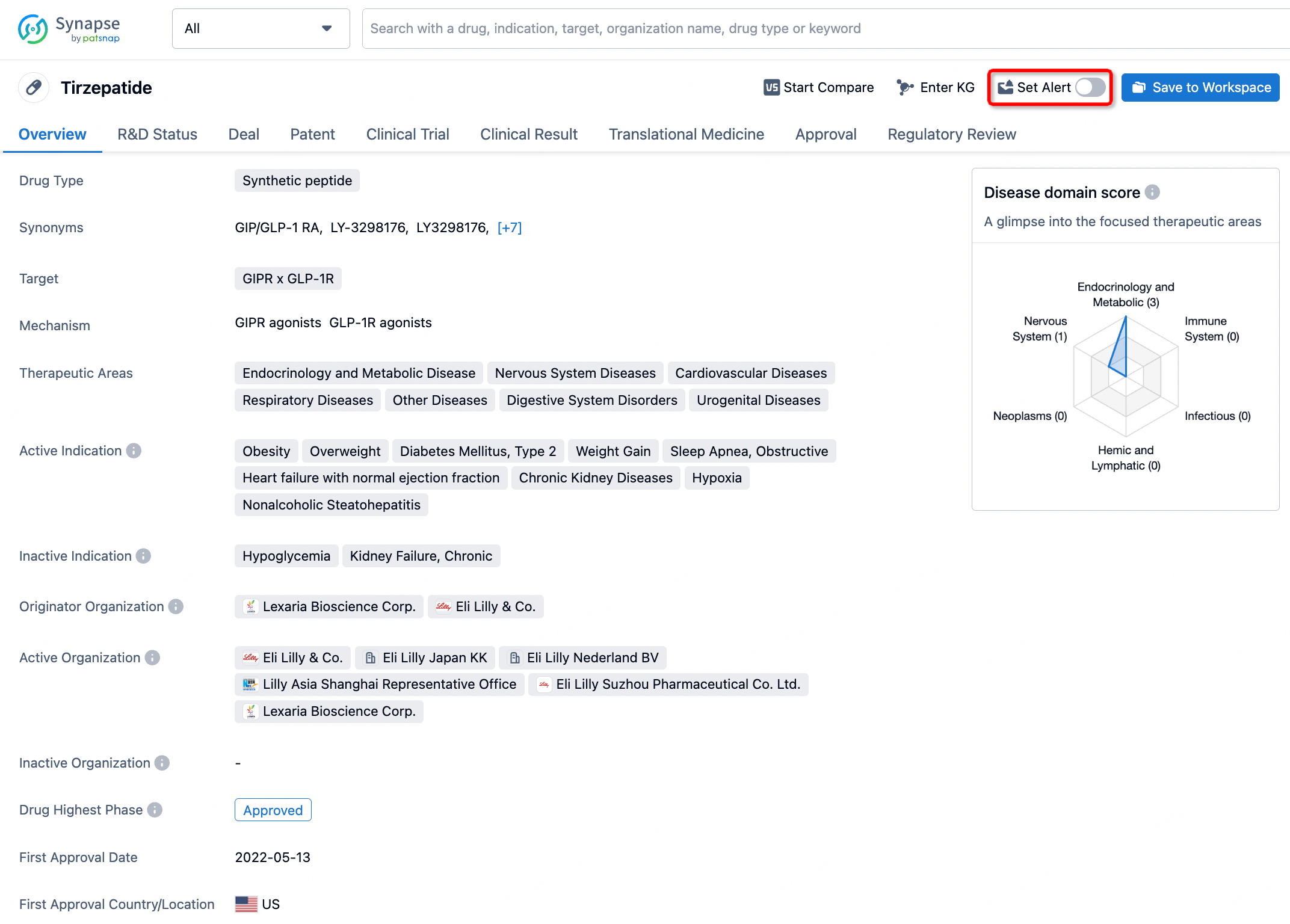
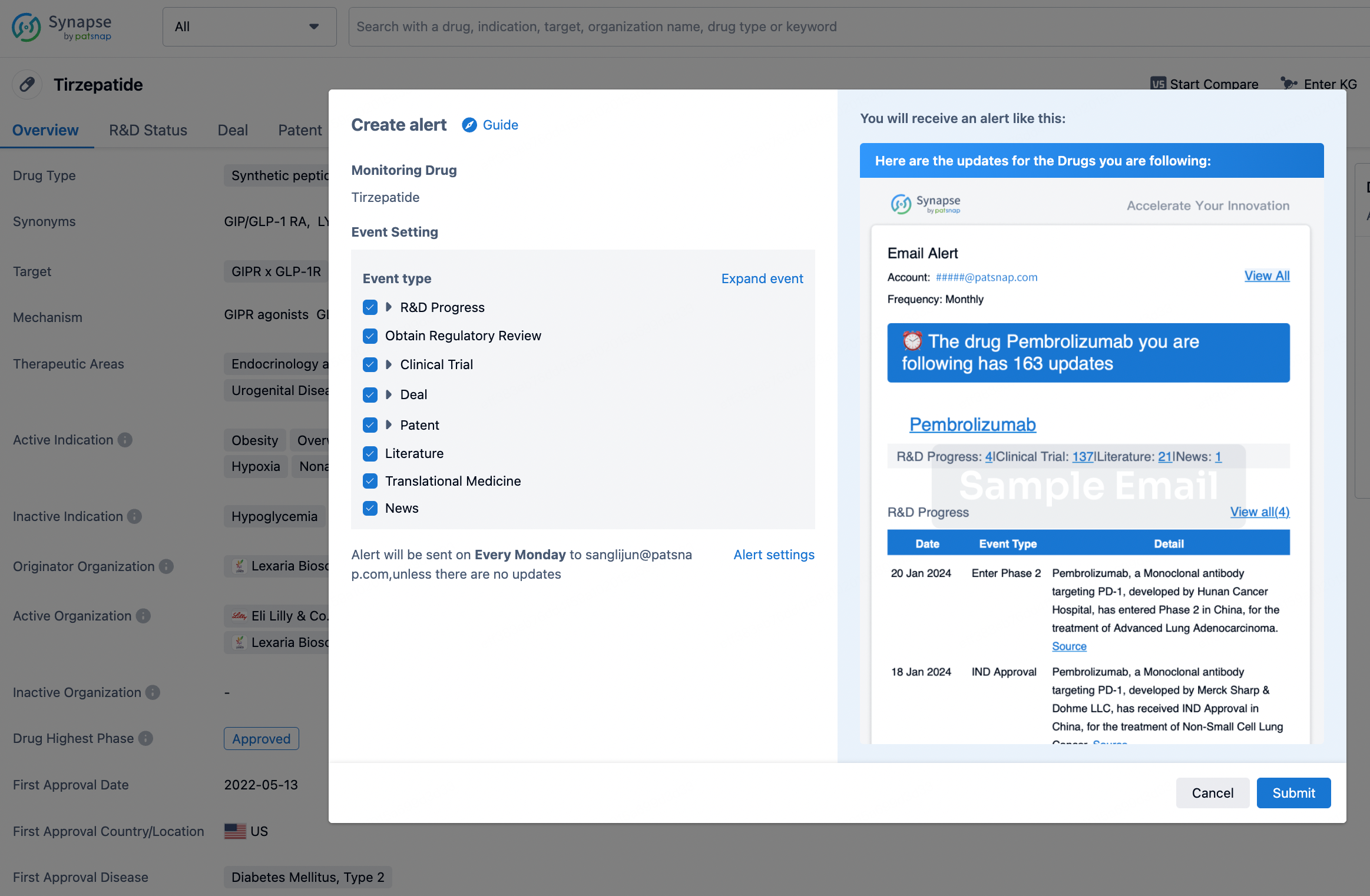
In the Synapse database, you can stay updated on the latest research and development advances of all drugs. This service is accessible anytime and anywhere, with updates available daily or weekly. Use the "Set Alert" function to stay informed. Click on the image below to embark on a brand new journey of drug discovery!
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.