Request Demo
What is Tralesinidase alfa used for?
28 June 2024
In recent years, advancements in biotechnology and pharmaceutical research have led to groundbreaking therapies for rare and previously untreatable conditions. One such advancement is Tralesinidase alfa, a novel enzyme replacement therapy (ERT) that has generated significant interest within the medical community. This biologic drug, produced through cutting-edge biotechnological processes, has demonstrated promising potential for the treatment of a specific lysosomal storage disorder. Researchers from various esteemed institutions have been closely studying Tralesinidase alfa, with ongoing clinical trials illuminating its efficacy and safety profile.
Tralesinidase alfa, an enzyme replacement therapy, specifically targets a rare genetic disorder known as mucopolysaccharidosis (MPS) type IIIB, also referred to as Sanfilippo syndrome type B. This disorder is caused by a deficiency in the enzyme alpha-N-acetylglucosaminidase (NAGLU), which is crucial for the breakdown and recycling of heparan sulfate, a complex sugar molecule found in the body. In individuals with MPS IIIB, the absence or malfunctioning of NAGLU leads to the accumulation of heparan sulfate within lysosomes, resulting in progressive cellular and tissue damage. The therapeutic goal of Tralesinidase alfa is to supplement the deficient enzyme, thereby reducing the build-up of heparan sulfate and mitigating the associated symptoms.
The development of Tralesinidase alfa has been spearheaded by several research institutions renowned for their contributions to rare disease therapeutics. These include biopharmaceutical companies specializing in enzyme replacement therapies and dedicated research organizations committed to advancing treatments for lysosomal storage disorders. Both preclinical studies and clinical trials have been conducted to evaluate the pharmacokinetics, pharmacodynamics, safety, and efficacy of Tralesinidase alfa. While it is still in the investigational stage, the data collected thus far has shown encouraging results, with patients experiencing notable improvements in clinical outcomes.
Tralesinidase alfa operates through a targeted mechanism of action designed to address the underlying enzymatic deficiency in MPS IIIB patients. The drug is a recombinant form of the human alpha-N-acetylglucosaminidase enzyme, produced using advanced genetic engineering techniques. Upon administration, Tralesinidase alfa is taken up by cells through receptor-mediated endocytosis, a process that allows the enzyme to enter lysosomes where it can exert its therapeutic effect.
Within the lysosomes, Tralesinidase alfa facilitates the degradation of accumulated heparan sulfate, thereby alleviating the pathological storage seen in MPS IIIB. By restoring the normal lysosomal function, Tralesinidase alfa helps prevent cellular and tissue damage, which in turn, can translate to clinical improvements in patients. Importantly, enzyme replacement therapies like Tralesinidase alfa are often administered intravenously, ensuring that the enzyme reaches systemic circulation and is distributed to various tissues and organs affected by the disease.
The primary indication for Tralesinidase alfa is the treatment of mucopolysaccharidosis type IIIB (MPS IIIB), a rare and debilitating lysosomal storage disorder. MPS IIIB is one of several subtypes of Sanfilippo syndrome, each characterized by deficiencies in different enzymes involved in the degradation of glycosaminoglycans. Patients with MPS IIIB typically present with a range of symptoms, including developmental delays, progressive cognitive decline, behavioral issues, sleep disturbances, and physical abnormalities. The disease often manifests in early childhood and progressively worsens over time, significantly impacting the quality of life and life expectancy of affected individuals.
By addressing the root cause of the enzyme deficiency in MPS IIIB, Tralesinidase alfa aims to slow down or halt disease progression, thereby improving patient outcomes. While traditional approaches to managing MPS IIIB have primarily focused on symptomatic relief and supportive care, the advent of Tralesinidase alfa represents a more targeted and potentially transformative treatment option. Clinical trials are ongoing to further elucidate the long-term benefits and potential risks of this therapy, with the hope of providing a much-needed therapeutic option for patients and families affected by this challenging condition.
In conclusion, Tralesinidase alfa embodies the promise of modern biotechnology in addressing rare genetic disorders. Its targeted mechanism of action and potential to improve the lives of those suffering from MPS IIIB highlight the importance of continued research and innovation in the field of enzyme replacement therapies. As clinical trials progress, the medical community remains hopeful that Tralesinidase alfa will soon become a pivotal treatment for MPS IIIB, offering new hope to patients and their families.
Tralesinidase alfa, an enzyme replacement therapy, specifically targets a rare genetic disorder known as mucopolysaccharidosis (MPS) type IIIB, also referred to as Sanfilippo syndrome type B. This disorder is caused by a deficiency in the enzyme alpha-N-acetylglucosaminidase (NAGLU), which is crucial for the breakdown and recycling of heparan sulfate, a complex sugar molecule found in the body. In individuals with MPS IIIB, the absence or malfunctioning of NAGLU leads to the accumulation of heparan sulfate within lysosomes, resulting in progressive cellular and tissue damage. The therapeutic goal of Tralesinidase alfa is to supplement the deficient enzyme, thereby reducing the build-up of heparan sulfate and mitigating the associated symptoms.
The development of Tralesinidase alfa has been spearheaded by several research institutions renowned for their contributions to rare disease therapeutics. These include biopharmaceutical companies specializing in enzyme replacement therapies and dedicated research organizations committed to advancing treatments for lysosomal storage disorders. Both preclinical studies and clinical trials have been conducted to evaluate the pharmacokinetics, pharmacodynamics, safety, and efficacy of Tralesinidase alfa. While it is still in the investigational stage, the data collected thus far has shown encouraging results, with patients experiencing notable improvements in clinical outcomes.
Tralesinidase alfa operates through a targeted mechanism of action designed to address the underlying enzymatic deficiency in MPS IIIB patients. The drug is a recombinant form of the human alpha-N-acetylglucosaminidase enzyme, produced using advanced genetic engineering techniques. Upon administration, Tralesinidase alfa is taken up by cells through receptor-mediated endocytosis, a process that allows the enzyme to enter lysosomes where it can exert its therapeutic effect.
Within the lysosomes, Tralesinidase alfa facilitates the degradation of accumulated heparan sulfate, thereby alleviating the pathological storage seen in MPS IIIB. By restoring the normal lysosomal function, Tralesinidase alfa helps prevent cellular and tissue damage, which in turn, can translate to clinical improvements in patients. Importantly, enzyme replacement therapies like Tralesinidase alfa are often administered intravenously, ensuring that the enzyme reaches systemic circulation and is distributed to various tissues and organs affected by the disease.
The primary indication for Tralesinidase alfa is the treatment of mucopolysaccharidosis type IIIB (MPS IIIB), a rare and debilitating lysosomal storage disorder. MPS IIIB is one of several subtypes of Sanfilippo syndrome, each characterized by deficiencies in different enzymes involved in the degradation of glycosaminoglycans. Patients with MPS IIIB typically present with a range of symptoms, including developmental delays, progressive cognitive decline, behavioral issues, sleep disturbances, and physical abnormalities. The disease often manifests in early childhood and progressively worsens over time, significantly impacting the quality of life and life expectancy of affected individuals.
By addressing the root cause of the enzyme deficiency in MPS IIIB, Tralesinidase alfa aims to slow down or halt disease progression, thereby improving patient outcomes. While traditional approaches to managing MPS IIIB have primarily focused on symptomatic relief and supportive care, the advent of Tralesinidase alfa represents a more targeted and potentially transformative treatment option. Clinical trials are ongoing to further elucidate the long-term benefits and potential risks of this therapy, with the hope of providing a much-needed therapeutic option for patients and families affected by this challenging condition.
In conclusion, Tralesinidase alfa embodies the promise of modern biotechnology in addressing rare genetic disorders. Its targeted mechanism of action and potential to improve the lives of those suffering from MPS IIIB highlight the importance of continued research and innovation in the field of enzyme replacement therapies. As clinical trials progress, the medical community remains hopeful that Tralesinidase alfa will soon become a pivotal treatment for MPS IIIB, offering new hope to patients and their families.
How to obtain the latest development progress of all drugs?
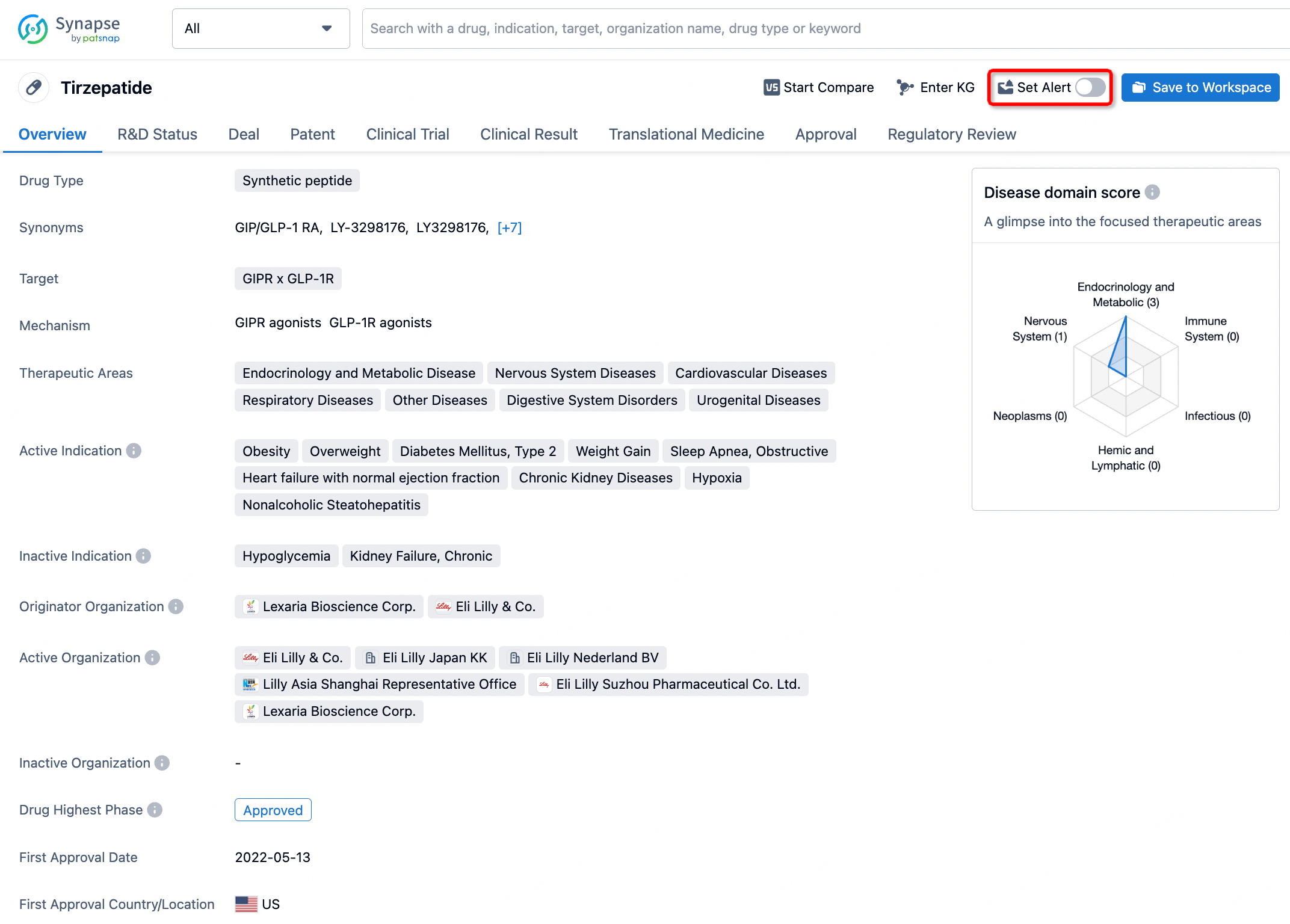
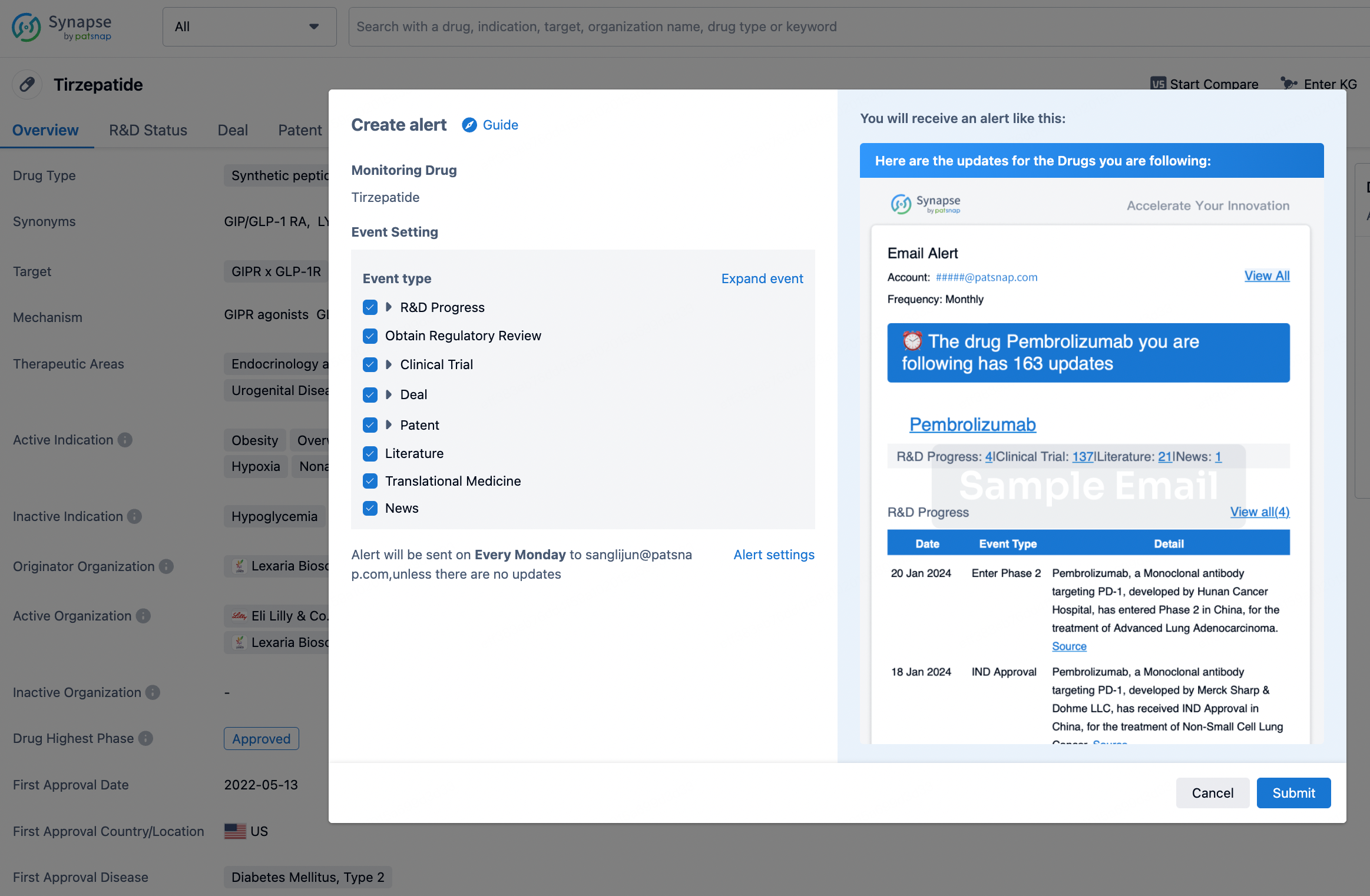
In the Synapse database, you can stay updated on the latest research and development advances of all drugs. This service is accessible anytime and anywhere, with updates available daily or weekly. Use the "Set Alert" function to stay informed. Click on the image below to embark on a brand new journey of drug discovery!
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.