Clinical Dose and Translational Science of the JAK1 Inhibitor Upadacitinib
Since its first approval in the United States in August 2019, Upadacitinib has rapidly scaled up to become a blockbuster drug within just two years. Sales reached $1.65 billion in 2021, $2.5 billion in 2022, and $1.604 billion in the first half of 2023, an increase of 51.7% year-over-year. With the approval of new indications and the expansion of its approved uses, AbbVie predicts that sales of Upadacitinib will exceed $7.5 billion by the year 2025.
Introduction to Upadacitinib
Upadacitinib was initially approved by the FDA and EMA in 2019 for the treatment of rheumatoid arthritis. Currently, upadacitinib has been approved in multiple markets for use in adults with Psoriatic Arthritis (PsA), Ankylosing Spondylitis (AS), non-radiographic axial spondyloarthritis, Crohn's Disease (CD), and Ulcerative Colitis (UC); as well as in adults and children aged 12 and over with Atopic Dermatitis (AD). Upadacitinib is also currently under evaluation for the treatment of other inflammatory diseases (NCT05843643, NCT04927975, NCT05889182, NCT06012240). Upadacitinib is available as a once-daily extended-release tablet, with dosages of 15, 30, and 45 mg in the United States, Europe, and many other countries.
👇Explore more about this drug by clicking the image below. Gain detailed insights into its R&D Status, Core Patent, Clinical Trials and Global Approval Status. Stay informed and updated.
Upadacitinib acts as an ATP-competitive JAK inhibitor; it competes with ATP and blocks nucleotide binding to inhibit kinase activity and the phosphorylation of downstream effectors. As a result, the formation of STAT dimers is prevented, which inhibits their translocation to the cell nucleus and binding to promoters. Enzyme activity assays indicate that upadacitinib has the strongest inhibitory effect on JAK1 (IC50 = 0.043 μM); it has weaker effects on JAK2 (IC50=0.12 μM), JAK3 (IC50 = 2.3 μM), and TYK2 (IC50 =4.7 μM). In cellular assays, upadacitinib demonstrates a selectivity for JAK1 over JAK2, JAK3, and TYK2 that is greater than 40-fold, 130-fold, and 190-fold, respectively.
Pharmacokinetic-pharmacodynamic analysis characterizing the relationship between upadacitinib plasma exposure and in vivo pharmacological effects shows that upadacitinib exerts stronger inhibition on IL-6 induced pSTAT3 (as a measure of JAK1 activity) compared to IL-7 induced pSTAT5 (as a measure of JAK1/3 activity). The potency ratio of IL-6 induced pSTAT3 to IL-7 induced pSTAT5 (based on the ratio of estimated EC50 values) is 2.0, consistent with the in vitro observation that upadacitinib has greater inhibitory effects on JAK1 compared to JAK3.
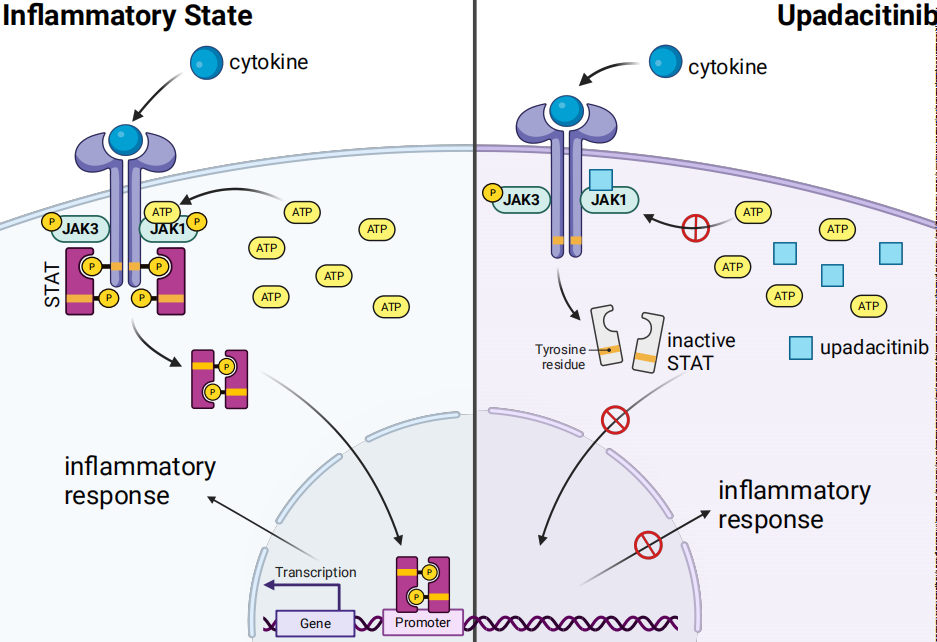
These findings support that upadacitinib effectively inhibits JAK1 while exhibiting weaker inhibition towards other subtypes, namely JAK2, JAK3, and TYK2. Since different receptors are associated with different JAKs, selective blockade of one JAK can suppress specific biological functions or target key cytokines involved in each type of immune-mediated inflammatory disease, while allowing other JAK-dependent cytokines to signal normally. Although upadacitinib demonstrates selectivity for JAK1, the enzymes of the JAK family work in concert, and as such, it may exert some biological effects on all pairings that involve JAK1.
The Pharmacokinetic/Pharmacodynamic Characteristics of Upadacitinib
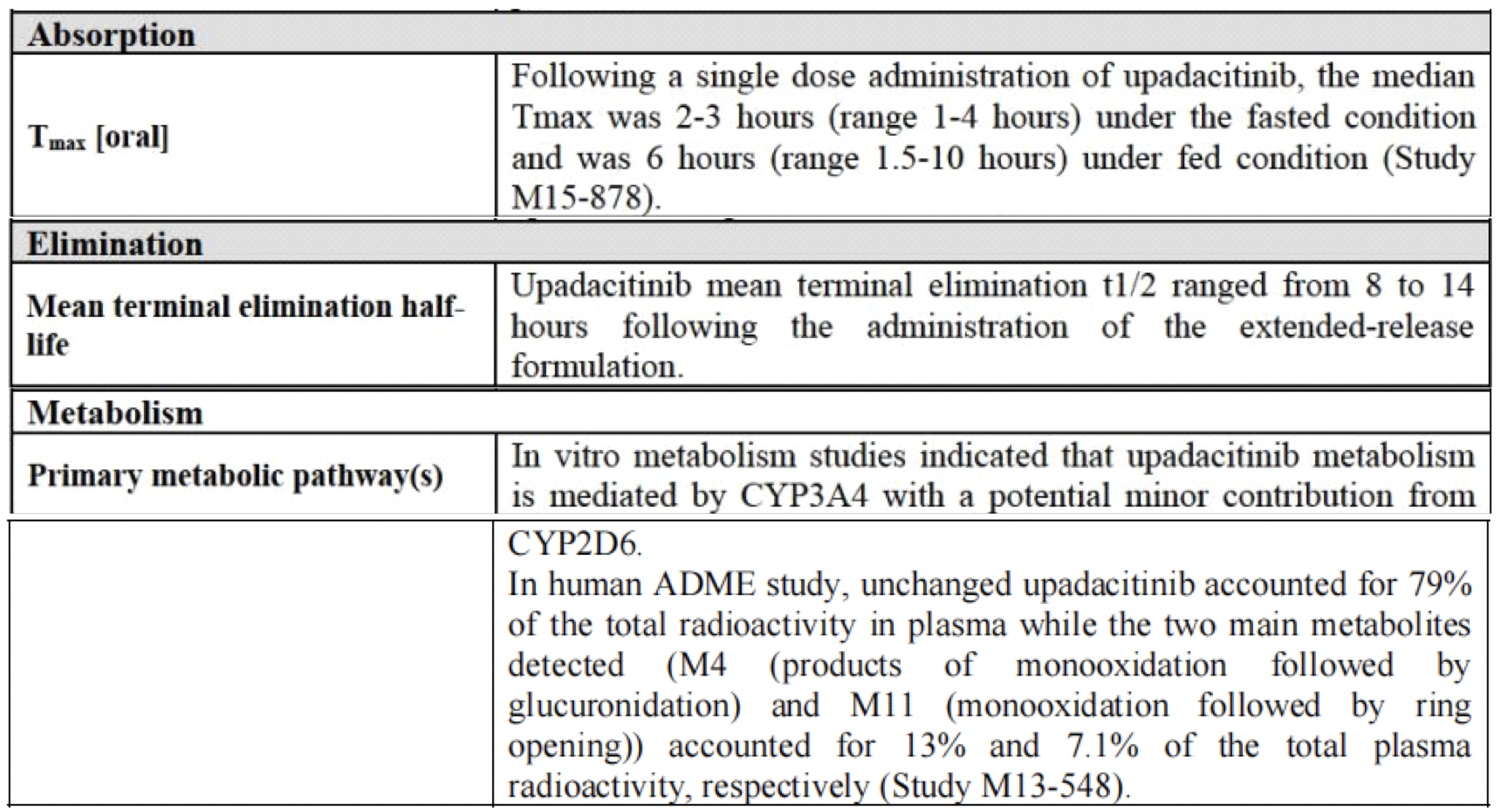
In humans, the plasma exposure of upadacitinib is proportional to the dose within the therapeutic dosage range. After oral administration of upadacitinib extended-release formulation, the median time to reach the maximum drug concentration (Tmax) in plasma is between 2 and 4 hours. With once-daily dosing of upadacitinib, steady-state plasma concentrations are achieved within 4 days, with minimal accumulation. The average terminal elimination half-life of upadacitinib is about 8 to 14 hours. Upadacitinib is primarily excreted as the parent drug in the urine (24%) and feces (38%), while the metabolites of upadacitinib account for approximately 34% of the drug's excretion. The pharmacological activity of upadacitinib is attributed to the parent molecule, which comprises 79% of the total plasma concentration, while the principal metabolite accounts for 13% of the total plasma concentration. No active metabolites of upadacitinib have been identified.
Compared to subjects with normal renal function (eGFR≥90ml/min/1.73 m^2), the changes in the average AUC_inf after a single dose of 15 mg upadacitinib were as follows: in subjects with mild (eGFR 60 to <90ml/min/1.73 m^2) renal impairment, there was an increase of 18%, and in those with moderate (eGFR 30 to <60ml/min/1.73 m^2) and severe (eGFR 15 to <30ml/min/1.73 m^2) renal impairment, increases were 33% and 44%, respectively. These changes in exposure levels do not necessitate a dose adjustment recommendation for patients with mild or moderate renal impairment. For patients with IBD (Inflammatory Bowel Disease) and severe renal impairment, the recommended induction and maintenance doses are 30mg QD and 15mg QD, respectively. For all other approved indications in patients with severe renal impairment, a recommended dose is 15mg daily. After a single dose of 15mg upadacitinib, the average AUC_inf in patients with mild and moderate hepatic impairment was increased by 28% and 24%, respectively, compared to patients with normal liver function. The average Cmax of upadacitinib remained unchanged in patients with mild hepatic impairment but was 43% higher in those with moderate hepatic impairment, compared to participants with normal liver function. Upadacitinib has not been studied in patients with severe hepatic impairment. Dose adjustment is not recommended for patients with mild or moderate hepatic impairment; this recommendation may vary by jurisdiction and recommended dosing should refer to the locally approved labeling.
The metabolism of upadacitinib is primarily mediated by CYP3A4, with a minor contribution from CYP2D6. Strong CYP3A inhibitors increase the AUC of upadacitinib by 75% and Cmax by 70%, while strong CYP3A inducers reduce the plasma exposure to upadacitinib by about half. The recommended dose of upadacitinib when coadministered with strong CYP3A inhibitors is 15mg QD, except for the induction dose for IBD, which is recommended to be 30mg QD. It is not advised to co-administer upadacitinib with strong CYP3A inducers, as a decrease in plasma exposure may lead to reduced therapeutic effects in patients. Concomitant use of strong CYP2D6 inhibitors or pH-modifying drugs does not affect the plasma exposure of upadacitinib.
Upadacitinib has been demonstrated to reversibly inhibit IL-6 and IL-7 cytokine signaling in a concentration-dependent manner in vivo, as assessed by IL-6-induced STAT3 phosphorylation (pSTAT3) and IL-7-induced STAT5 phosphorylation (pSTAT5) in vitro. The temporal profiles of these signaling events caused by upadacitinib follow plasma concentration levels, with maximum inhibition coinciding with peak plasma concentrations, and STAT phosphorylation levels returning to near baseline by the end of the dosing interval.
These pharmacokinetic-pharmacodynamic analyses were utilized for dose selection in the initial phase 2 studies conducted throughout the development program of upadacitinib.
Clinical Efficacy of Upadacitinib
The efficacy of upadacitinib compared to placebo or active comparators has been evaluated in clinical trials across different approved immune-mediated inflammatory diseases. In all clinical trials, upadacitinib achieved the primary endpoints and most secondary endpoints, demonstrating superiority over placebo in the background of standard care therapies for RA (Rheumatoid Arthritis), PsA (Psoriatic Arthritis), AS (Ankylosing Spondylitis), nr-axSpA (non-radiographic axial Spondyloarthritis), AD (Atopic Dermatitis), UC (Ulcerative Colitis), and CD (Crohn's Disease). Furthermore, upadacitinib was superior to Adalimumab and Abatacept in the context of RA, to Dupilumab in the context of AD, and non-inferior to Adalimumab in the context of PsA. These results underscore the robustness of upadacitinib's clinical efficacy. The effectiveness and safety of upadacitinib in the real-world setting have also been corroborated.
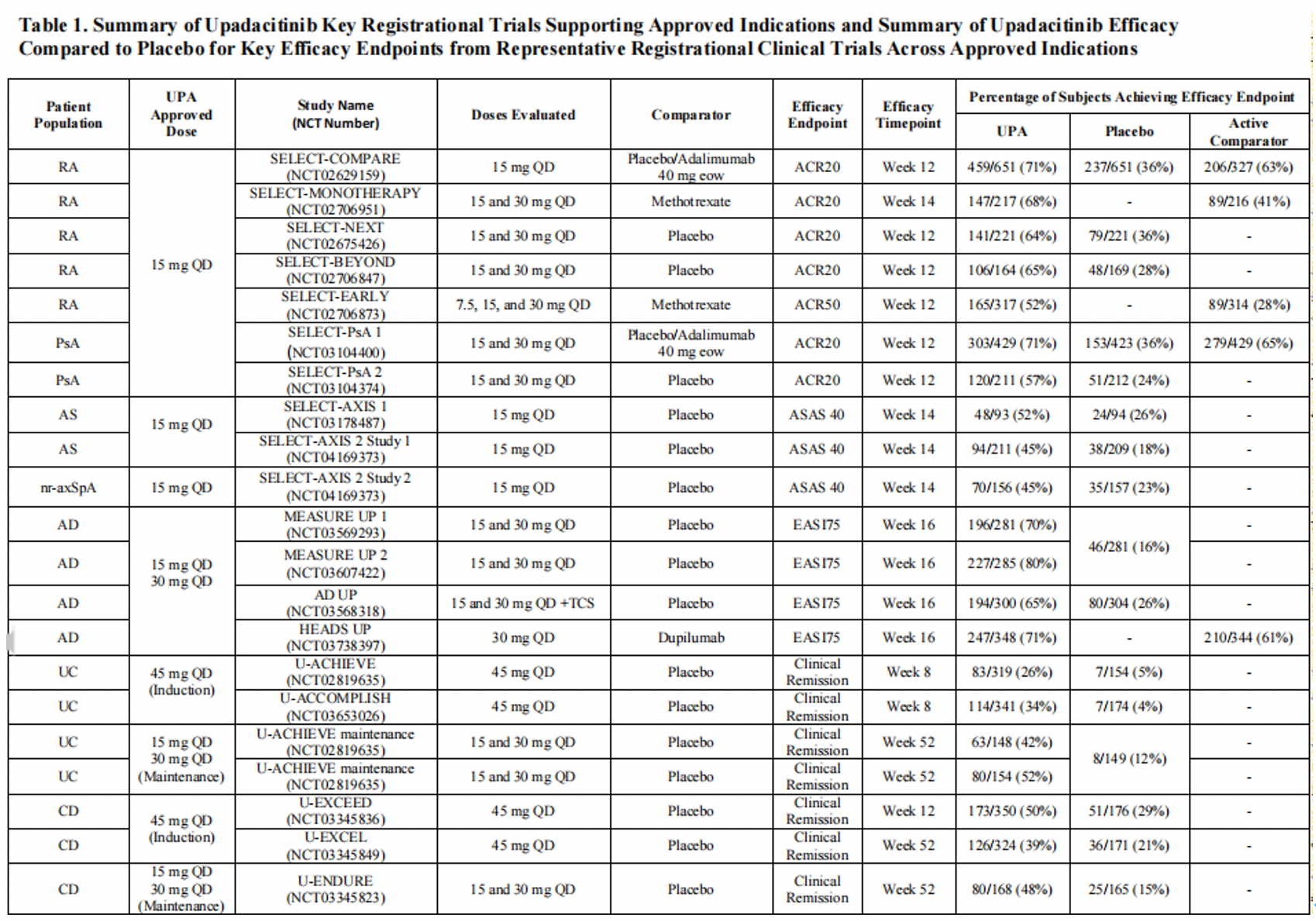
The relationship between upadacitinib plasma exposure and efficacy varies with different clinical conditions, leading to approved dosages tailored for various immune-mediated inflammatory diseases. This variability may be driven by differences in the inflammatory burden, sites of inflammation, and associated cytokine pathways among the diseases. Exposure-response relationships between the mean plasma concentration (C_avg) of upadacitinib and selected efficacy endpoints have been established. For RA and PsA, the efficacy of upadacitinib plasma exposure under a 15 mg QD dosing regimen approaches a plateau at approximately median C_avg of 15 ng/mL. On the other hand, increased plasma exposure of upadacitinib does not show a notable trend in enhancing response rates, suggesting that plasma exposure at 15 mg QD maximizes the response. Analysis of UC and CD induction data indicates that the efficacy of upadacitinib is maximized when exposed to a 45 mg QD regimen (approximately median C_avg of 40 ng/mL) at the end of the induction period. For atopic dermatitis as well as maintenance of UC and CD, a 30 mg QD exposure (approximately median C_avg of 29 ng/mL) provides additional benefits in chronic use compared to the 15 mg QD regimen, leading to both dosages being approved for these indications. Furthermore, dose recommendations for special populations (such as patients with renal or hepatic impairment) or for different indications using strong CYP3A inhibitors or inducers are based on exposure ranges assessed in pivotal phase III trials, as well as the exposure-response relationships of efficacy and safety for each indication.
The Clinical Safety of Upadacitinib
The safety of upadacitinib has been assessed in a comprehensive clinical development program, demonstrating consistent safety across indications such as RA (rheumatoid arthritis), PsA (psoriatic arthritis), axSpA (axial spondyloarthritis), UC (ulcerative colitis), CD (Crohn's disease), and AD (atopic dermatitis). Upadacitinib generally exhibited good tolerability, with observed safety differences potentially reflecting the diverse patient characteristics in different populations and comorbidities that impact background risk factors. The most common adverse events reported in clinical trials of upadacitinib were similar and included upper respiratory tract-related infections, nasopharyngitis, and elevated levels of creatine phosphokinase, with acne being more frequently reported in AD trials.
According to Burmester et al., the pattern, characteristics, and incidence of COVID-19 infections observed in patients treated with upadacitinib, including the frequency of events and the frequency leading to hospitalization, were generally similar to those seen in the general population. The incidence of malignancies (excluding non-melanoma skin cancer) did not show significant changes over time with exposure to different diseases and dosages of upadacitinib. In RA and PsA populations, the rates of herpes zoster (HZ), NMSC (non-melanoma skin cancer), and increased creatine phosphokinase levels were higher with upadacitinib compared to adalimumab. The majority of HZ events on upadacitinib were mild or moderate in severity, rarely led to discontinuation of therapy, and involved a single dermatome without cases involving the central nervous system or internal organs. The incidence of NMSC with upadacitinib was generally consistent across different diseases and was not observed in AS. The NMSC incidence was slightly higher in the 30 mg upadacitinib group compared to the 15 mg group in AD. NMSC events were generally not severe and did not lead to treatment discontinuation. Deaths, serious infections, major adverse cardiovascular events (MACE), venous thromboembolism, and malignancies were observed, with the lowest incidence rates in AS and AD; there is no evidence to suggest that mortality rates in RA, PsA, or AD patients exposed to upadacitinib are higher than expected in the general population. The number of deaths reported in the IBD program was too limited to be compared with the general population, and no deaths were reported in the AS program.
Summary
A post-hoc analysis assessing the safety of upadacitinib in patients with a high cardiovascular disease risk from the SELECT-RA clinical program, including the SELECT-COMPARE trial, which was a head-to-head study comparing upadacitinib with adalimumab.
The analysis indicated that, compared with the overall population, there was an increased risk of MACE (major adverse cardiovascular events), malignancies (excluding non-melanoma skin cancer, NMSC), and venous thromboembolism in high-risk patients, yet the incidence of these events was comparable between patients treated with upadacitinib and adalimumab. Across all cohorts, a higher incidence of NMSC and herpes zoster (HZ) was observed in the upadacitinib group, and an increased rate of serious infections was also detected in upadacitinib-treated patients with higher cardiovascular risk. While the interpretation of this post-hoc analysis may be associated with certain limitations, the data might also suggest that upadacitinib has distinctive characteristics compared to other JAK inhibitors.
Overall, the safety profile of upadacitinib has been well-characterized in a broad range of immune-mediated inflammatory diseases, and the aforementioned efficacy results support its favorable benefit-risk profile for use in its approved indications.

References
1.Mohamed MF et.al; Upadacitinib: Mechanism of action, clinical and translational science.Clin Transl Sci. 2023 Nov 20. doi: 10.1111/cts.13688.
2.Huang IH et.al;.JAK-STAT signaling pathway in the pathogenesis of atopic dermatitis: An updated review. Front Immunol. 2022;13:1068260.