Science: AlphaFold2 Structural Models Guide Drug Discovery
On May 16th, researchers led by Professor Brian K. Shoichet from the University of California, San Francisco, and Professor Bryan L. Roth from the University of North Carolina at Chapel Hill, published a research paper titled "AlphaFold2 Structures Guide Prospective Ligand Discovery" in the journal Science.

The researchers conducted a prospective docking of extensive compound libraries against the unoptimized AlphaFold2 (AF2) models of the σ2 receptor and the 5-HT2A receptor. They tested hundreds of new molecules and compared the results to those obtained from docking against experimental structures. High and comparable hit rates and affinities were observed for both experimental structures and AF2 models.
Cryo-electron microscopy was used to determine the structure of an efficient 5-HT2A ligand identified in the AF2 docking. This revealed adaptive residue adjustments similar to those predicted by AF2. Although AF2 models may capture conformations different from experimental structures, these conformations are still low-energy states and are crucial for ligand discovery. This expands the scope of structure-based ligand discovery.
Research Background
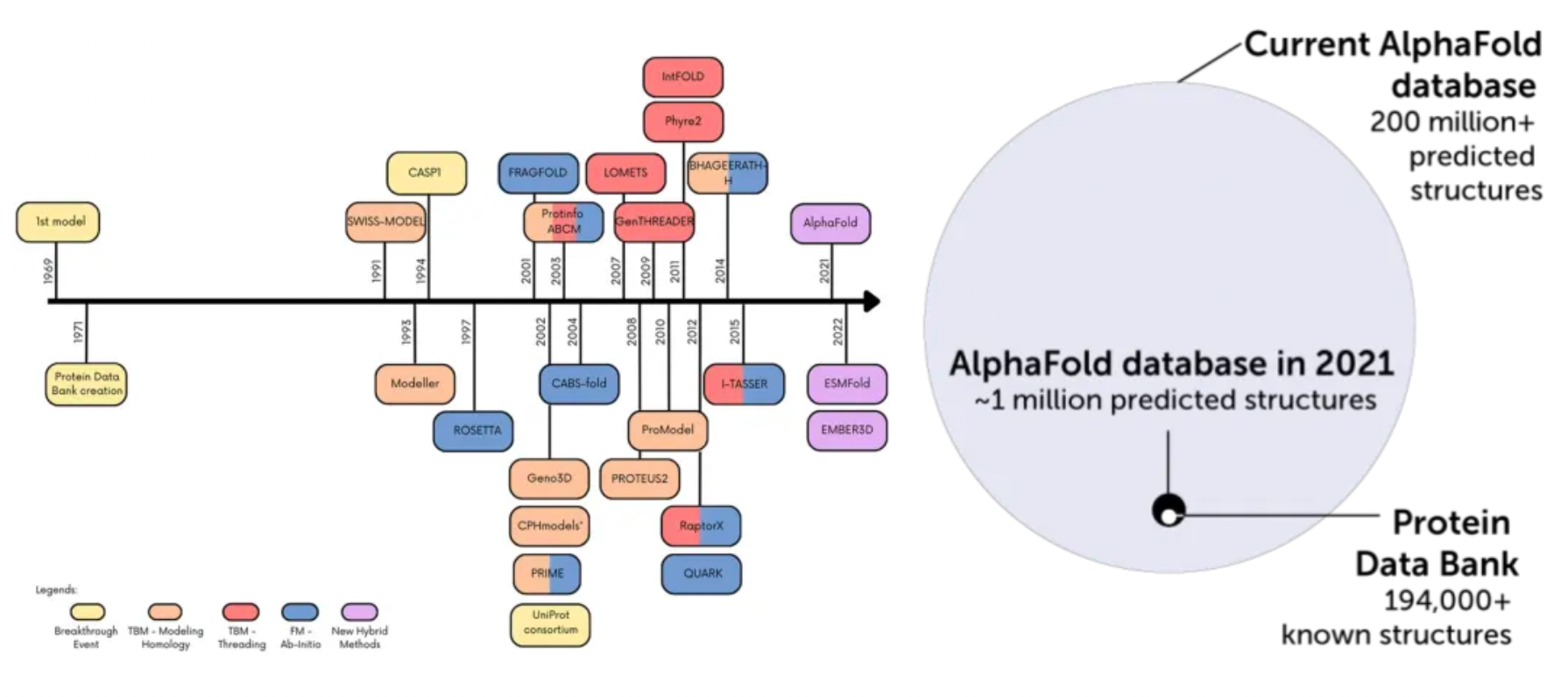
AlphaFold2's breakthroughs in protein structure prediction have demonstrated unprecedented capabilities, enabling atomic-level accuracy in structural predictions and scalable operations. As of now, the AlphaFold database contains protein structure information of the human proteome and proteins from 47 other organisms, encompassing over 200 million proteins and nearly all potential therapeutic protein targets. These structures have shown immense value in applications such as structural biology, protein design, protein-protein interactions, target prediction, protein function prediction, and the study of biological mechanisms.
However, the impact of AF2 structures on structure-based ligand discovery remains unclear. In ligand binding sites, high fidelity to low-energy conformations is considered critical. Even relatively minor errors, which might be inconsequential for other applications, can interfere with ligand recognition and conformation prediction. Retrospective studies suggest that docking experiments with unoptimized AF2 models struggle with identifying known ligands and predicting their conformations compared to benchmark molecules. However, they perform better in direct comparisons with experimental structures. A drawback of such studies is their susceptibility to historical bias: known ligands may influence the conformations taken by experimental structures when determining complex structures, and experimental structures may, in turn, affect the process of new ligand exploration.
Research Protocol
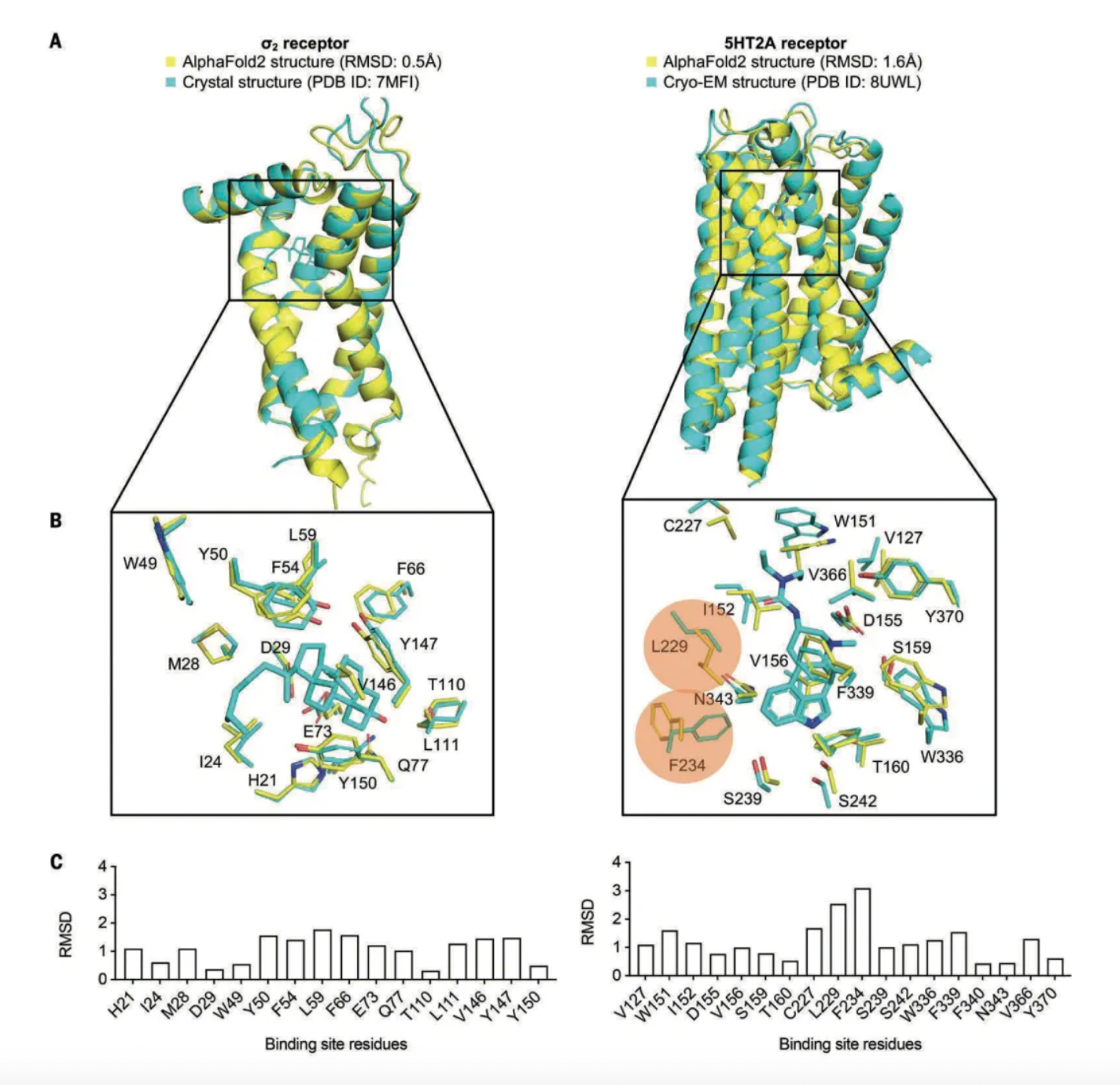
First, the researchers compared the AF2 model's structural prediction of the σ2 receptor (EXPERA) and the 5-HT2A receptor (GPCRs). Because the AF2 model made these predictions before the crystal structures of these receptors were published, this helps to minimize potential bias.
For the σ2 receptor, the AF2 model accurately predicted the side-chain conformations of all positive site residues, with a root mean square deviation (RMSD) from the crystal structure of only 1.1 Å. Only a few residues had individual RMSD values greater than 1.5 Å. Regarding the 5-HT2A receptor, while most binding site residues were well predicted (with RMSD from the cryo-EM structure being less than 2 Å), two residues had RMSDs ranging from 2.5 to 3.1 Å, adopting different rotamers.
Subsequently, the researchers used DOCK3.8 software to sample millions of potential ligand poses and scored each molecular pose based on physical fitness, typically targeting a predefined and fixed binding site. In DOCK3.8, the fit of ligands to the protein is assessed by calculating two types of interactions: electrostatic interactions using a probe charge model based on the Poisson-Boltzmann equation, and van der Waals interactions using the AMBER force field, with corrections for ligand desolvation and conformational strain.
For the crystal structure of the σ2 receptor, the researchers previously screened 490 million dockable molecules from the ZINC20 database. Among the top 300,000 ranked molecules (0.06% of the ranked database), 138 high-scoring molecules were selected for radioligand competition assays, of which 70 displaced over 50% of the known ligand [³H]-DTG at a concentration of 1μM—a hit rate of 51%. The best 21 active molecules had Ki values ranging from 1.8 nM to low μM, with 19 having Ki < 50 nM, and 6 having Ki < 5 nM.
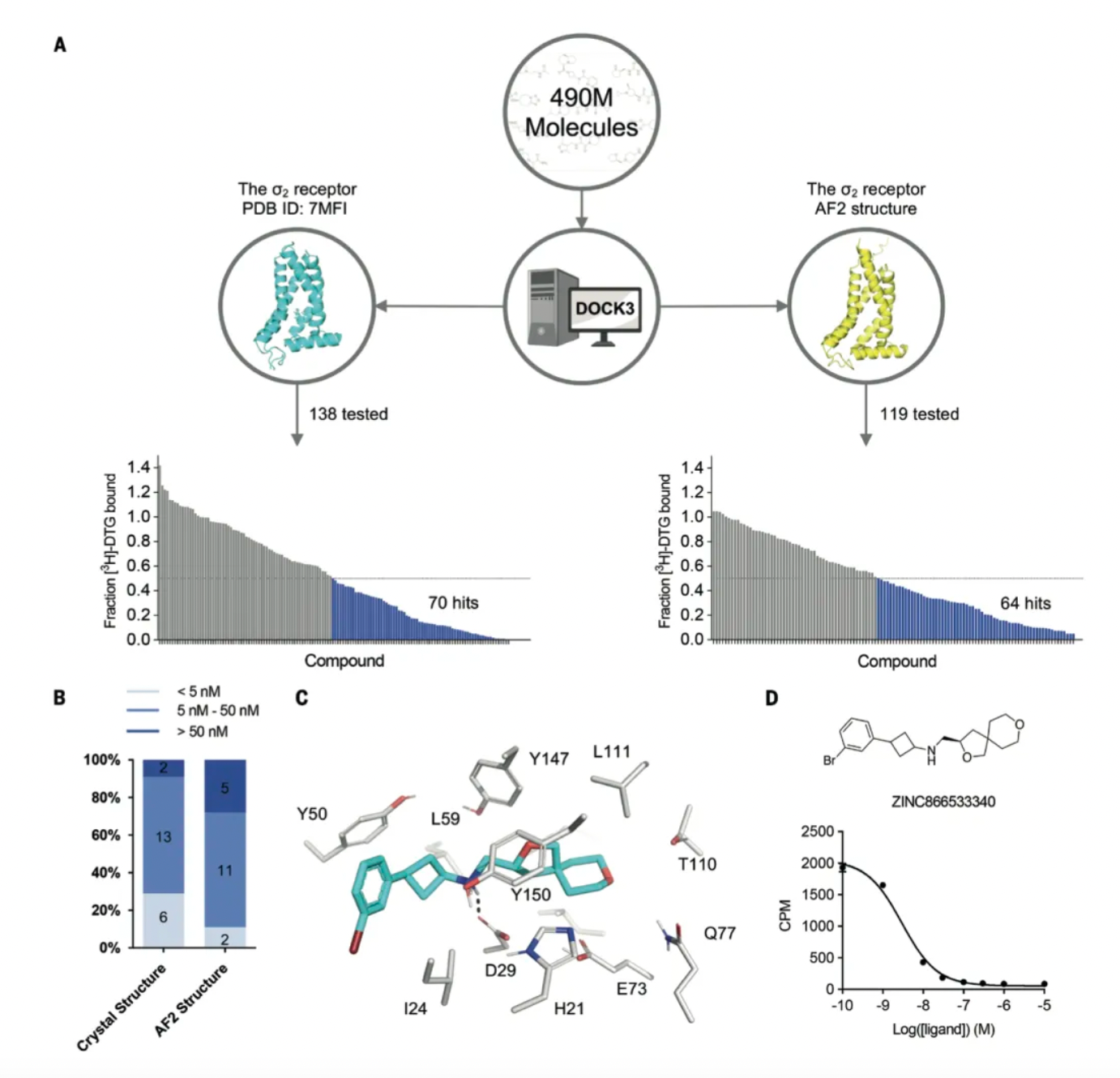
Meanwhile, the researchers docked the same library of 490 million molecules against the AF2 model of the σ2 receptor and selected 119 new molecules from the top 30,000 dockings for synthesis and testing. Of these, 64 molecules displaced more than 50% of specific [³H]-DTG σ2 binding at 1 μM concentration, resulting in a hit rate of 54%. Although this rate is slightly higher than the 51% observed in the crystal structure docking test, there was no significant difference between the two hit rates based on a z-test. The Ki values of the top 18 hits from the AF2 test ranged from 1.6 nM to 84 nM, with 13 having Ki values less than 50 nM and 2 less than 5 nM.
When the researchers redocked the 490 million molecules to the σ2 AF2 model, the previously high-ranking 138 molecules dropped in rank to such an extent that they no longer appeared within the top 30,000. This likely reflects a slight contraction in the AF2 positive site compared to the crystal structure, with the computed volume reducing from 268 ų to 213 ų.
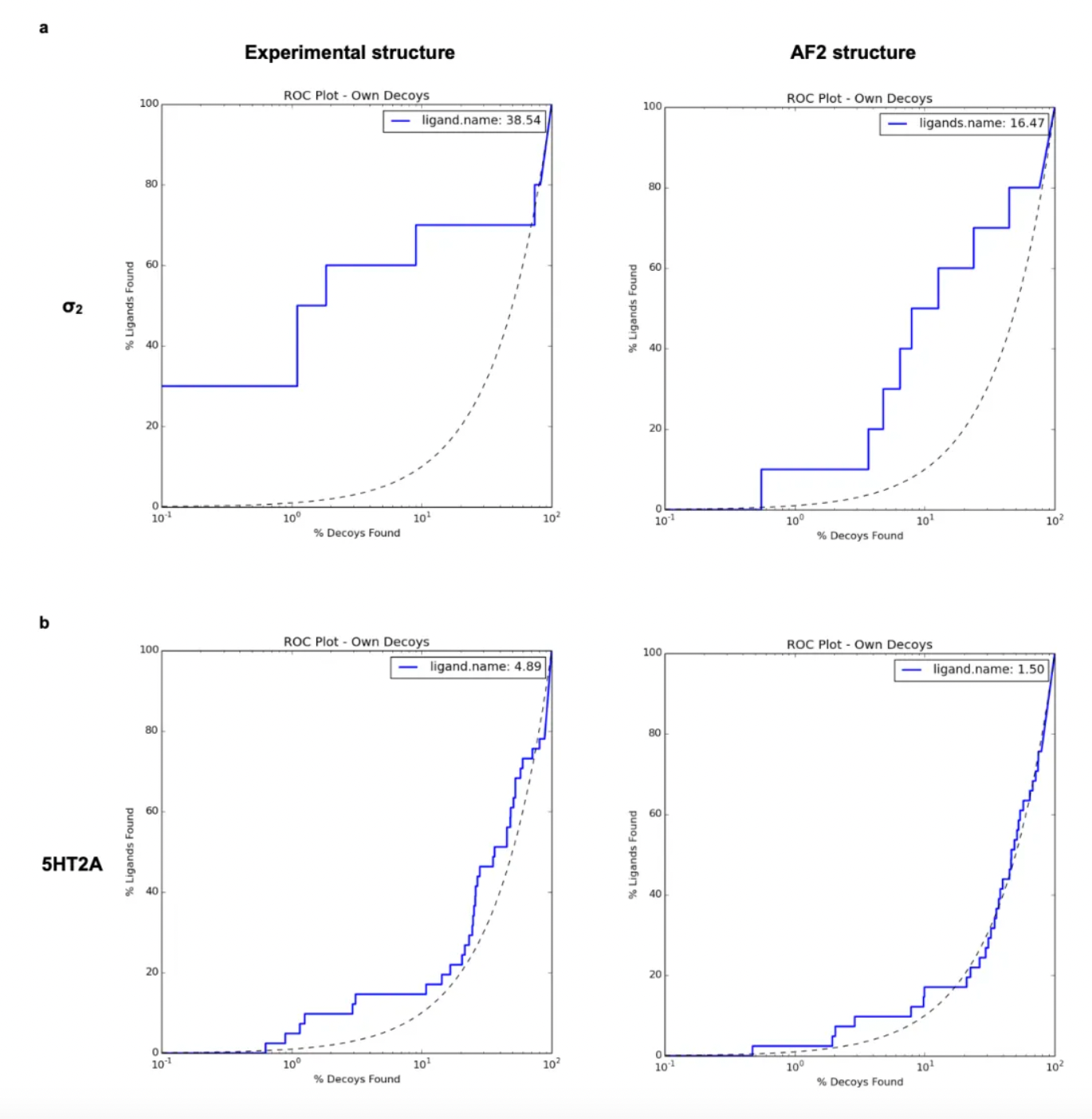
Correspondingly, when using the crystal structure (logAUC: 39) versus the AF2 model (logAUC: 16) to dock known σ2 ligands from the ChEMBL database along with a property-matched decoy library, the crystal structure yielded higher enrichment. Similar results have been observed in retrospectives involving cryo-EM and AF2 structures of the 5-HT2A receptor: experimental structures outperformed AF2 structures in the retrospective enrichment of known ligands.
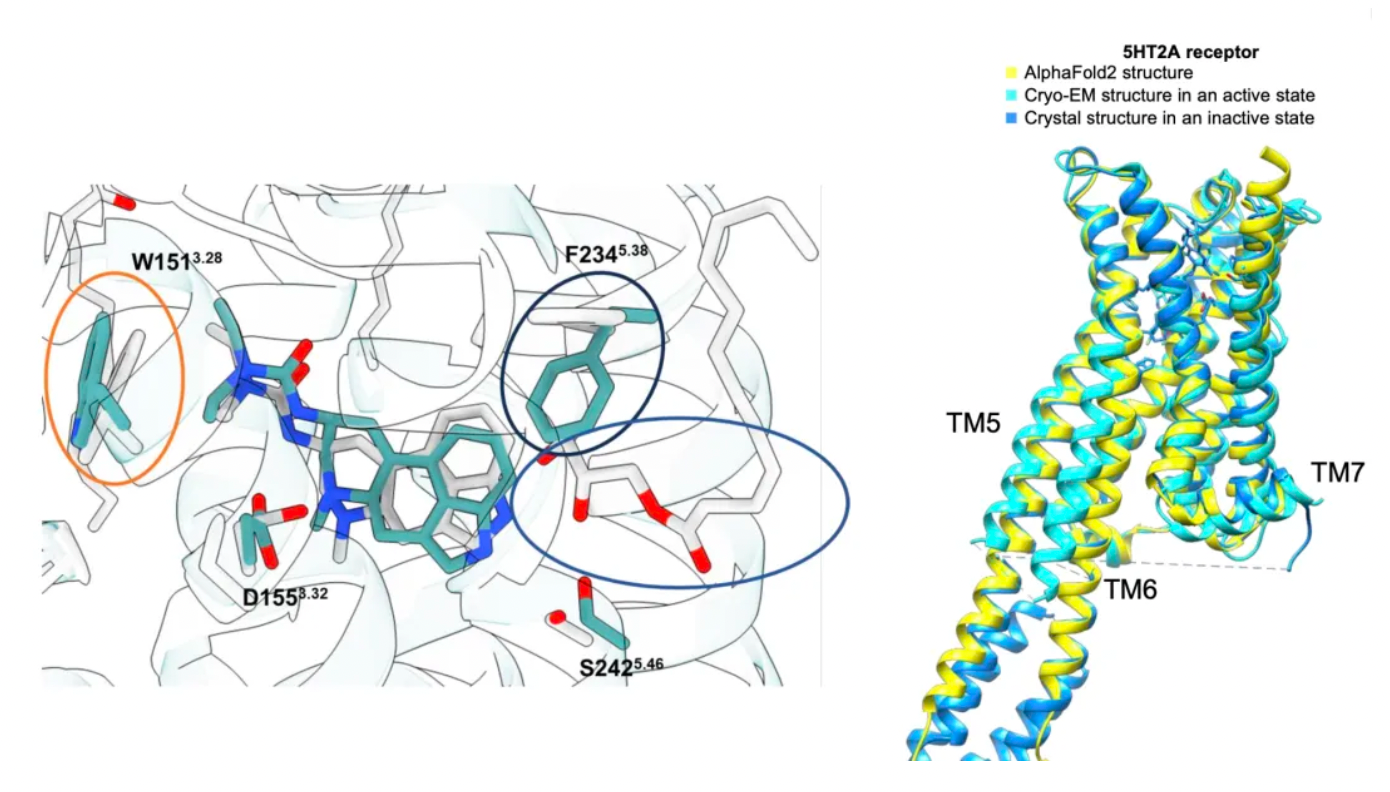
Unlike the situation with the σ2 receptor, the AF2 model of the 5-HT2A receptor shows significant rotameric variations at several key residues compared to the cryo-EM structure. Although the RMSD (Root-Mean-Square Deviation) of most key residues is less than 1.5 Å, critical residues such as Phe-2345.38 on TM5 and Leu-22945.52 in the ECL2 loop show deviations of 2.5 Å and 3.1 Å, respectively, and adopt different rotamers in the two structures. This difference likely arises because the AF2 model leverages the overall sequence similarity within the 5-HT receptor family, whereas the cryo-EM structure represents the active state of the receptor in complex with Gαq, while the AF2 model is closer to the inactive state. Researchers expect that the experimentally determined structure is more likely to select agonists with 5-HT2A selectivity and Gαq bias.
Subsequently, researchers docked over 1.6 billion molecules into both the cryo-EM and AF2 receptor structures. They considered the top 3 million molecules (top 0.2%) for each structure based on their docking scores. Consistent filtering, clustering, and hit selection criteria were applied to these molecules to narrow down to a set of priority compounds for synthesis and testing. Ultimately, 223 high-ranking molecules resulting from docking to the cryo-EM structure and 161 high-ranking molecules from docking to the AF2 structure were synthesized.
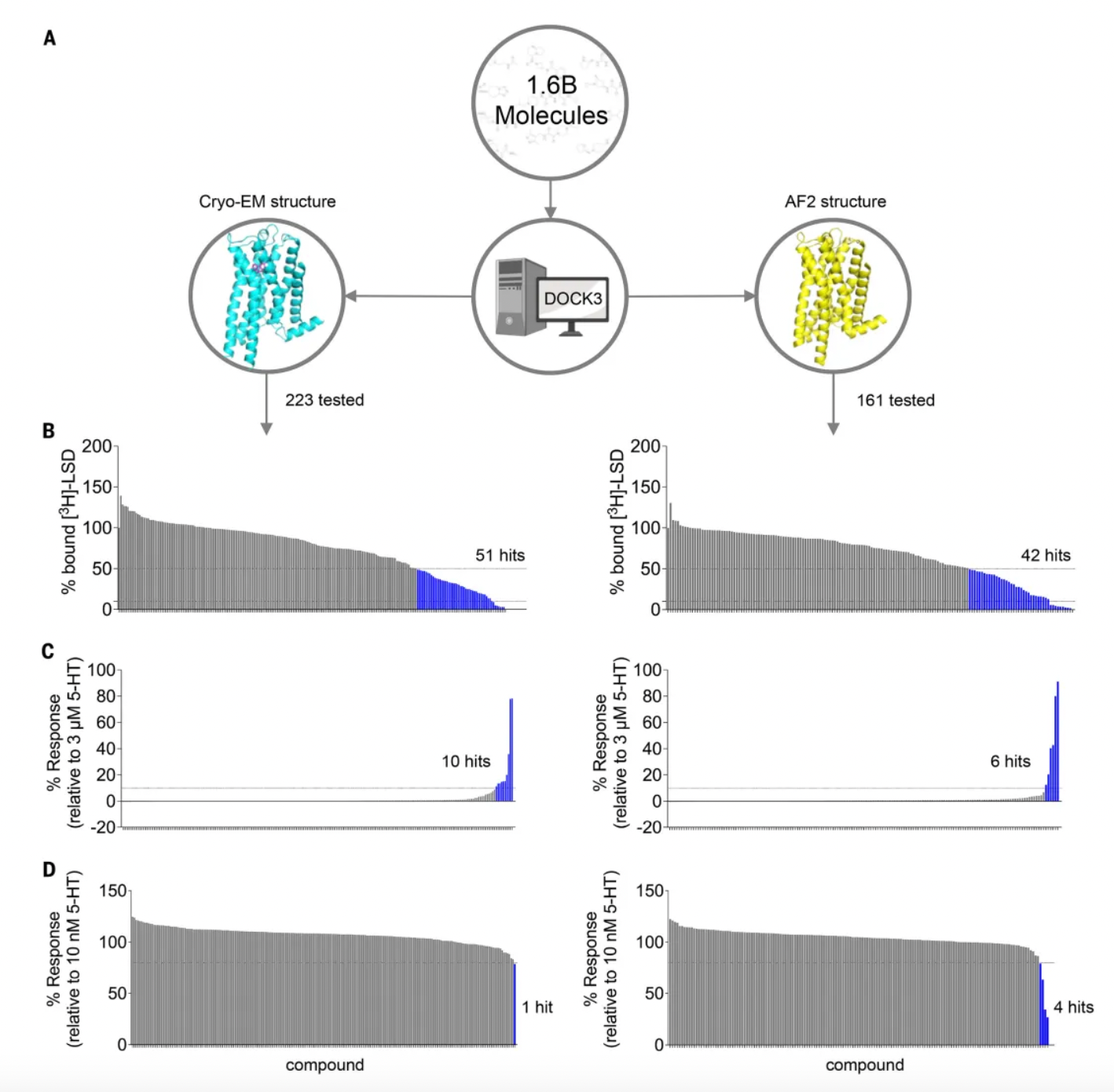
In preliminary radioligand binding screening assays, out of 223 molecules docked against cryo-EM structures, 51 substituted more than 50% of [3H]-LSD at a docking ligand concentration of 10 μM, with a hit rate of 23%. Additionally, out of 161 molecules docked against AF2 models, 42 met this threshold, resulting in a hit rate of 26%. Unexpectedly, the three highest affinity compounds (15 to 24 nM) all came from AF2 docking, whereas the three best compounds from cryo-EM docking had affinities approximately five times weaker (ranging from 71 to 114 nM). Despite the higher hit rate from AF2 docking, there was no significant difference in hit rates between AF2 and experimental docking based on the z-test. Moreover, among the 93 active molecules common to both tests, no two shared the same scaffold, with an average pairwise similarity (Tc) of 0.27, akin to randomness.
Given that the 5-HT receptor has multiple subtypes in the human body, to evaluate the subtype selectivity of the molecules, researchers assessed the functional activity (including agonist or antagonist activity) of 338 priority molecules from AF2 and experimental docking. This included subtype selectivity across 5-HT2A, 5-HT2B, and 5-HT2C receptors, as well as ligand bias between Gαq activation or β-arrestin2 recruitment.
Upon treatment with 3 μM of each compound, 10 compounds from the cryo-EM docking group and 6 compounds from the AF2 docking group exhibited activity as 5-HT2A agonists, defined as ≥10% of the 5-HT response; additionally, another 10 compounds (5 cryo-EM and 5 AF2) and 6 compounds (3 cryo-EM and 3 AF2) were identified as 5-HT2B and 5-HT2C agonists, respectively. In antagonist screening mode at 3 μM, 5 compounds (1 cryo-EM and 4 AF2) antagonized 5-HT2A receptor activity, defined as ≥20% of Clozapine activity. Furthermore, 17 compounds (10 cryo-EM and 7 AF2) and 1 compound (cryo-EM) were discovered to have antagonist activity on 5-HT2B and 5-HT2C receptors, respectively.
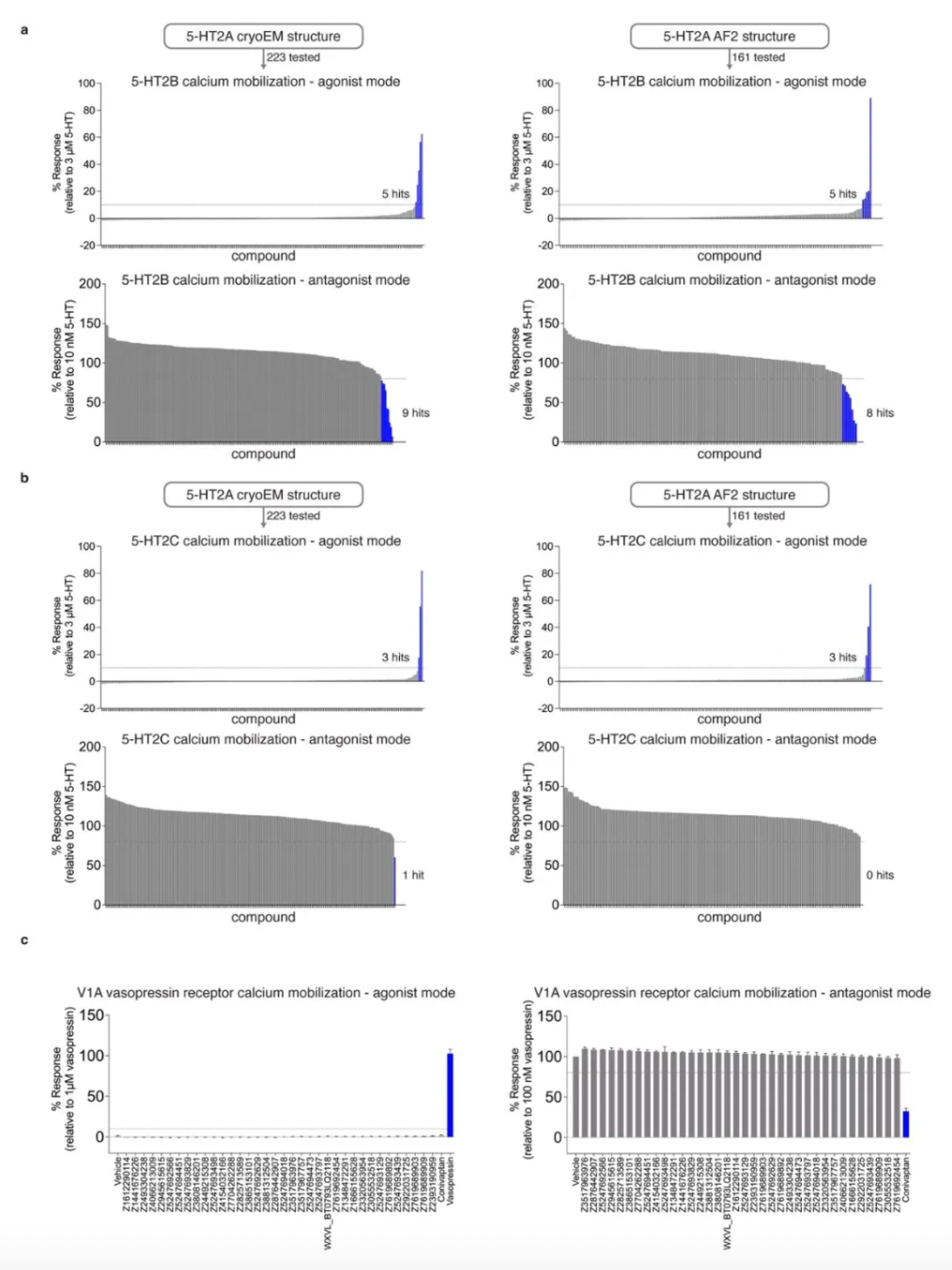
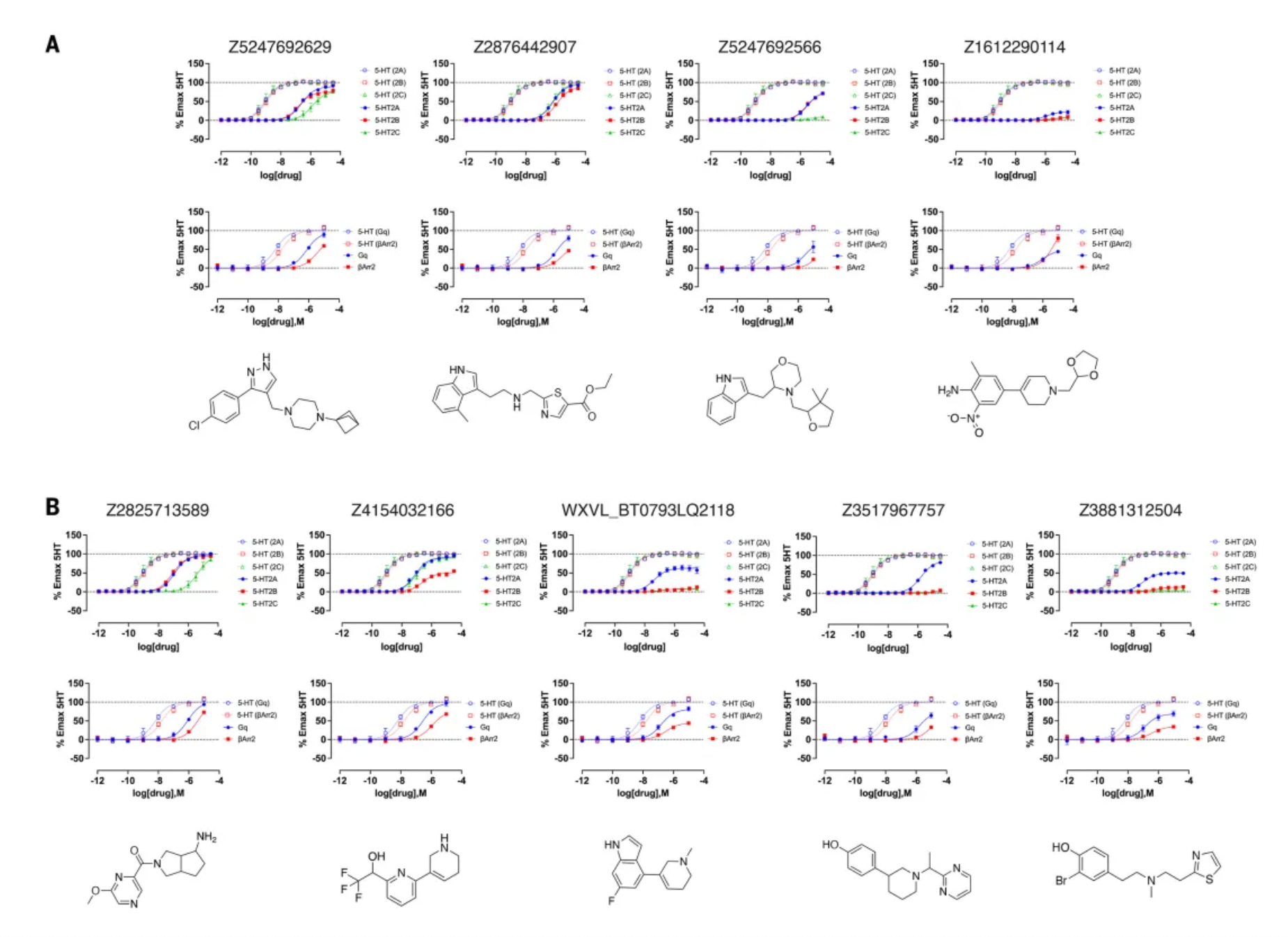
Further activity studies revealed that the potency (pEC50 values) of top agonists from the cryo-EM docking group ranged from 246 nM to 3 µM, while those from the AF2 docking group ranged from 42 nM to 1.6 µM. Among the top five AF2 agonists, three (Q2118, Z7757, and Z2504) exhibited subtype selectivity for the 5-HT2A receptor over the 5-HT2B and 5-HT2C receptors. In contrast, none of the top cryo-EM agonists demonstrated selectivity for the 5-HT2A receptor. During comprehensive dose-response studies, the AF2-derived agonists emerged as the most effective and selective molecules.
Research Summary
Contrary to expectations, the study results indicate that large library docking using the AF2 model is equally effective as docking using experimental structures. For the σ2 and 5-HT2A receptors, hundreds of molecules were experimentally tested on both structures, yielding a high hit rate with no significant differences between the AF2 model and the experimental structures. Comparisons of the affinity of these molecules led to similar conclusions. Specifically, for the 5-HT2A receptor, compounds generated using the AF2 structure might even be more potent and selective, with docking hits showing sub-nanometer EC50 values that rank among the most potent and selective molecules in extensive structure-based docking for this target. Cryo-EM structures validated the model predictions for the novel agonist Z7757: the docking predictions overlapped well with the new cryo-EM structure positions, and some key structural differences between the original AF2 model and the lisuride/5-HT2A cryo-EM structure were reproduced in the new Z7757/5-HT2A complex's cryo-EM structure. These observations are consistent with the view that the AF2 model captures low-energy, accessible states of the 5-HT2A receptor.
In conclusion, the authors propose that alternative conformations sampled by the AF2 model capture low-energy conformations, which are useful for identifying ligands different from those identified by the experimental structures. In docking tests against the 5-HT2A receptor, the functional properties of hits from AF2 docking were not inferior to those from experimental structure docking. Notably, three 5-HT2A receptor hits derived from AF2 model docking were subtype-selective, a feature not observed in docking against the cryo-EM structures. The AF2 model can expand the range of proteins targeted by structure-based drug discovery and broaden the spectrum of effective chemical types discovered.
How to obtain the latest research advancements in the field of biopharmaceuticals?
In the Synapse database, you can keep abreast of the latest research and development advances in drugs, targets, indications, organizations, etc., anywhere and anytime, on a daily or weekly basis. Click on the image below to embark on a brand new journey of drug discovery!