Request Demo
Last update 10 May 2025
BIWH-3(C.H. Boehringer Sohn AG & Co. KG)
Last update 10 May 2025
Overview
Basic Info
Drug Type Recombinant protein |
Synonyms pyro-Glu-rhMCP-1, BIWH 3, BIWH-3 + [1] |
Target |
Action modulators |
Mechanism CCL2 modulators(C-C motif chemokine 2 modulators) |
Therapeutic Areas |
Active Indication- |
Inactive Indication |
Originator Organization |
Active Organization- |
Inactive Organization |
License Organization- |
Drug Highest PhaseDiscontinuedPhase 1 |
First Approval Date- |
Regulation- |
Login to view timeline
Related
2
Clinical Trials associated with BIWH-3(C.H. Boehringer Sohn AG & Co. KG)NCT02249117
Pharmacokinetics and Pharmacodynamics of BIWH 3: a Randomised, Placebo-controlled, Double Blind Dose Escalation Study (0.02, 0.06, 0.2, 0.6, and 2.0 μg/kg Intravenous Over One Hour) in Healthy Duffy Positive vs. Duffy Negative Male Volunteers
NCT02215824
A Randomised, Double-blind, Placebo-controlled, Dose Escalation Study to Investigate Safety, Pharmacokinetics and Pharmacodynamics of Different Doses (0.2, 0.6, 2.0, 6.0, and 20.0 μg/hr) of BIWH 3 Administered for 6 Hours in Patients With Chronic Critical Limb Ischaemia (CLI, Fontaine Class III or IV). COINART-1 Trial (First COllateral INto ARTery Trial)
100 Clinical Results associated with BIWH-3(C.H. Boehringer Sohn AG & Co. KG)
Login to view more data
100 Translational Medicine associated with BIWH-3(C.H. Boehringer Sohn AG & Co. KG)
Login to view more data
100 Patents (Medical) associated with BIWH-3(C.H. Boehringer Sohn AG & Co. KG)
Login to view more data
19
Literatures (Medical) associated with BIWH-3(C.H. Boehringer Sohn AG & Co. KG)01 Oct 2018Bioorganic & medicinal chemistry lettersQ4 · MEDICINE
Inhibition of the LOX enzyme family members with old and new ligands. Selectivity analysis revisited
Q4 · MEDICINE
Article
Author: Major, Balázs ; Lőrincz, Zsolt ; Hajdú, István ; Fabó, Gabriella ; Dormán, György ; Kardos, József ; Cseh, Sándor
Lysyl oxidase (LOX) enzymes as potential drug targets maintain constant attention in the therapy of fibrosis, cancer and metastasis. In order to measure the inhibitory activity of small molecules on the LOX enzyme family members a fluorometric activity screening method was developed. During assay validation, previously reported non-selective small inhibitor molecules (BAPN, MCP-1, thiram, disulfiram) were investigated on all of the major LOX enzymes. We confirmed that MCP-1, thiram, disulfiram are in fact pan-inhibitors, while BAPN inhibits only LOX-like enzymes (preferably LOX-like-protein-2, LOXL2) in contrast to the previous reports. We measured the LOX inhibitory profile of a small targeted library generated by 2D ligand-based chemoinformatics methods. Ten hits (10.4% hit rate) were identified, and the compounds showed distinct activity profiles. Potential inhibitors were also identified for LOX-like-protein-3 (LOXL3) and LOX-like-protein-4 (LOXL4), that are considered as emerging drug targets in the therapy of melanoma and gastric cancer.
01 Oct 2013The journal of painQ2 · MEDICINE
Induction of Monocyte Chemoattractant Protein-1 (MCP-1) and Its Receptor CCR2 in Primary Sensory Neurons Contributes to Paclitaxel-Induced Peripheral Neuropathy
Q2 · MEDICINE
Article
Author: Yan Li ; Patrick M. Dougherty ; Haijun Zhang ; Seo Yeon Yoon ; Edgar T. Walters ; Alyssa K. Kosturakis ; Jessica A. Boyette-Davis
PERSPECTIVE:
CIPN is a severe side effect accompanying paclitaxel chemotherapy and lacks effective treatments. The current study suggests that blocking MCP-1/CCR2 signaling could be a new therapeutic strategy to prevent or reverse paclitaxel CIPN. This preclinical evidence encourages future clinical evaluation of this strategy.
01 Oct 2013NeuropharmacologyQ2 · MEDICINE
Monocyte Chemoattractant Protein-1 upregulates GABA-induced current: Evidence of modified GABAA subunit composition in cortical neurons from the G93A mouse model of Amyotrophic Lateral Sclerosis
Q2 · MEDICINE
Article
Author: Pieri, Massimo ; Guglielmotti, Angelo ; Zona, Cristina ; Antonini, Alessia ; Caioli, Silvia ; Severini, Cinzia
Amyotrophic Lateral Sclerosis (ALS) is a neurodegenerative disorder that affects upper and lower motor neurons. Previous evidence has indicated that excitotoxic cell death in ALS may remarkably depend on Cl(-) ion influx through the GABA(A) receptors. In this study we have analysed the effect of Monocyte Chemoattractant Protein-1 (MCP-1), a chemokine expressed to a higher level in ALS patients, on GABAA receptors in cultured cortical neurons from a genetic model of ALS (G93A) and compared with wild type SOD1 (SOD1) and their corresponding non transgenic littermates (Control). By performing electrophysiological experiments we have observed that, in cortical neurons MCP-1 (2-150 ng/ml) induced an enhancement of GABA-evoked currents that was significantly higher in G93A neurons compared to controls. The effect of MCP-1 was not dependent on the activation of its receptor CCR2, while it was blocked by flumazenil, the antagonist of benzodiazepine sites. Analysis of GABAA receptor subunit composition has indicated an altered subunit expression level in G93A cortical neurons compared to controls. Instead, in cultured spinal neurons MCP-1 induced a significant reduction of GABA-evoked currents, also through the benzodiazepine sites, indicating a region-specific mechanism of action. However, no differences were observed in the current reduction between the three neuronal populations. These findings provide the first evidence that MCP-1, acting on benzodiazepine sites, can modulate the GABA-evoked currents, depending on the subunit composition of GABA(A) receptor. In cortical neurons MCP-1 upmodulates the GABA-evoked current and this effect is exacerbated in the mutated neurons. It is reasonable to assume that the higher Cl(-) influx through GABA(A) receptors in the presence of MCP-1 in mutated cortical neurons may induce an excitotoxicity acceleration. Agents able to block the MCP-1 production may then prove useful for ALS treatment.
100 Deals associated with BIWH-3(C.H. Boehringer Sohn AG & Co. KG)
Login to view more data
R&D Status
10 top R&D records. to view more data
Login
| Indication | Highest Phase | Country/Location | Organization | Date |
|---|---|---|---|---|
| Chronic Limb-Threatening Ischemia | Phase 1 | - | 01 Oct 2002 |
Login to view more data
Clinical Result
Clinical Result
Indication
Phase
Evaluation
View All Results
Not Applicable | - | (ADPKD patients) | sghbzkxdih(dotcanjrpe) = ijqymvjbph qjkhrocwaz (sezdzkytlc ) | - | 29 May 2021 | ||
(Controls) | sghbzkxdih(dotcanjrpe) = kiyfymaavm qjkhrocwaz (sezdzkytlc ) |
Login to view more data
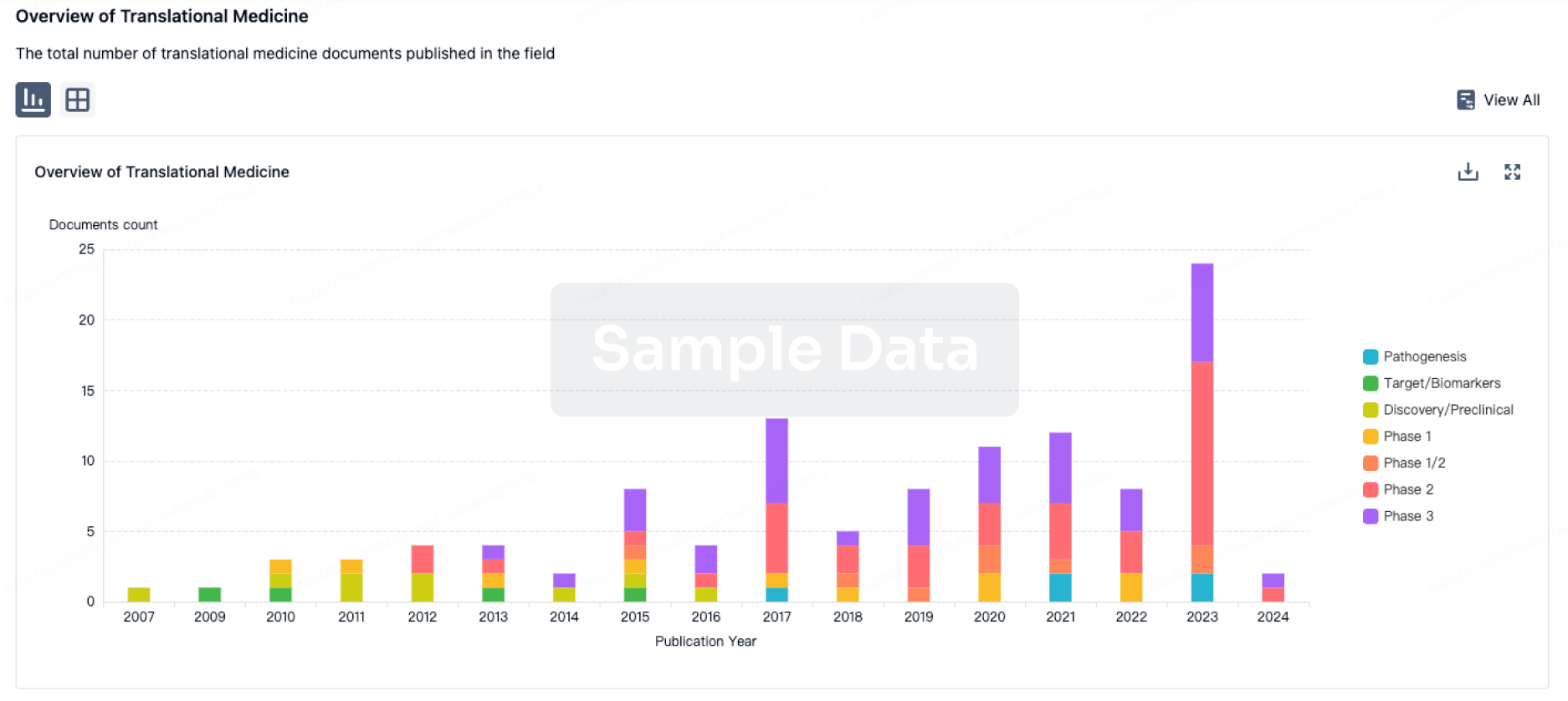
Translational Medicine
Boost your research with our translational medicine data.
login
or
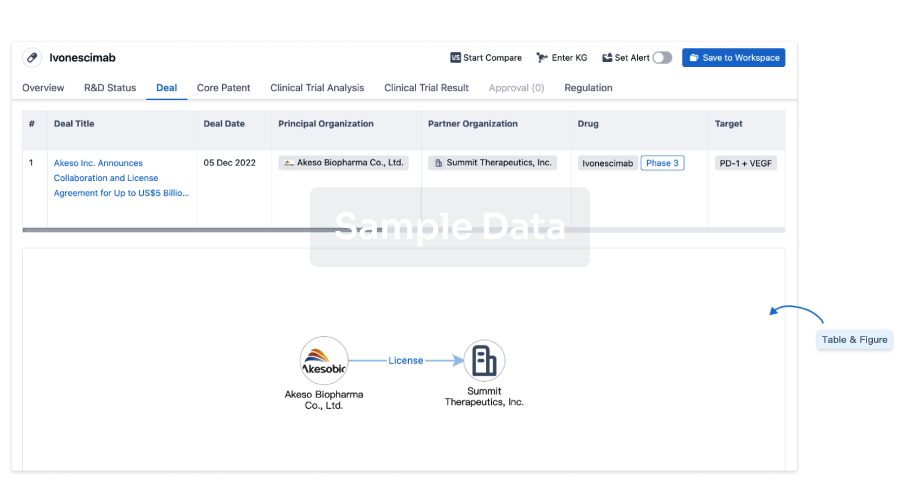
Deal
Boost your decision using our deal data.
login
or
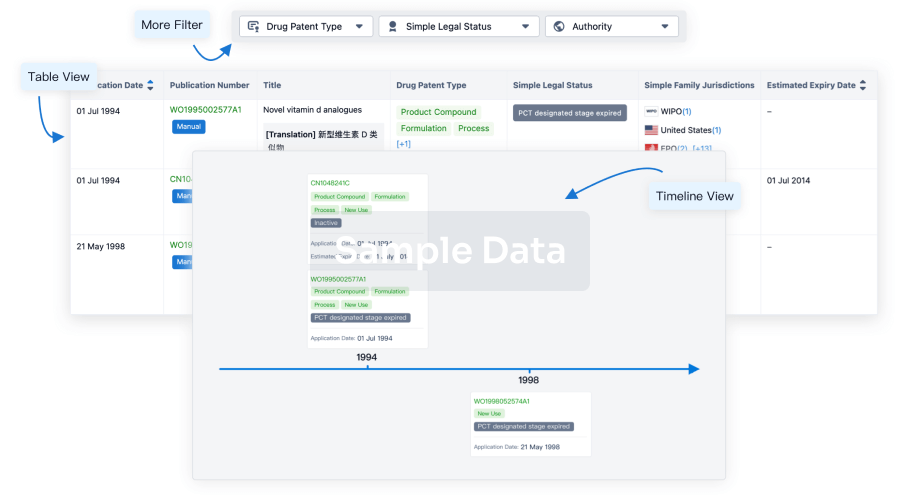
Core Patent
Boost your research with our Core Patent data.
login
or
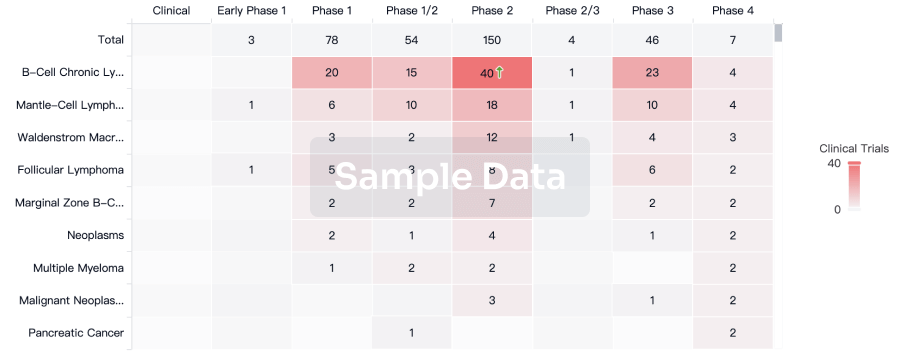
Clinical Trial
Identify the latest clinical trials across global registries.
login
or
Approval
Accelerate your research with the latest regulatory approval information.
login
or
Biosimilar
Competitive landscape of biosimilars in different countries/locations. Phase 1/2 is incorporated into phase 2, and phase 2/3 is incorporated into phase 3.
login
or

Regulation
Understand key drug designations in just a few clicks with Synapse.
login
or
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.
Bio
Bio Sequences Search & Analysis
Sign up for free
Chemical
Chemical Structures Search & Analysis
Sign up for free