Request Demo
Last update 20 Oct 2025
Demethylmenaquinone methyltransferase
Last update 20 Oct 2025
Basic Info
Synonyms- |
Introduction- |
Related
Target |
Mechanism |
Active Org. |
Originator Org. |
Active Indication |
Inactive Indication |
Drug Highest Phase |
First Approval Ctry. / Loc. |
First Approval Date |
100 Clinical Results associated with Demethylmenaquinone methyltransferase
Login to view more data
100 Translational Medicine associated with Demethylmenaquinone methyltransferase
Login to view more data
Login to view more data
01 Feb 2020Nature chemistryQ1 · CHEMISTRY
Repurposing human kinase inhibitors to create an antibiotic active against drug-resistant Staphylococcus aureus, persisters and biofilms
Q1 · CHEMISTRY
Article
Author: Hackl, Mathias W ; Korotkov, Vadim S ; Lehmann, Johannes ; Mandl, Franziska A M ; Le, Philipp ; Kuster, Bernhard ; Sieber, Stephan A ; Reinecke, Maria ; Pieper, Dietmar H ; Hacker, Stephan M ; Medina, Eva ; Kunold, Elena ; Antes, Iris ; Wuest, William M ; Fetzer, Christian ; Ugur, Ilke ; Chaves-Moreno, Diego ; Rohde, Manfred ; Macsics, Robert ; Rox, Katharina ; Jennings, Megan C
New drugs are desperately needed to combat methicillin-resistant Staphylococcus aureus (MRSA) infections. Here, we report screening commercial kinase inhibitors for antibacterial activity and found the anticancer drug sorafenib as major hit that effectively kills MRSA strains. Varying the key structural features led to the identification of a potent analogue, PK150, that showed antibacterial activity against several pathogenic strains at submicromolar concentrations. Furthermore, this antibiotic eliminated challenging persisters as well as established biofilms. PK150 holds promising therapeutic potential as it did not induce in vitro resistance, and shows oral bioavailability and in vivo efficacy. Analysis of the mode of action using chemical proteomics revealed several targets, which included interference with menaquinone biosynthesis by inhibiting demethylmenaquinone methyltransferase and the stimulation of protein secretion by altering the activity of signal peptidase IB. Reduced endogenous menaquinone levels along with enhanced levels of extracellular proteins of PK150-treated bacteria support this target hypothesis. The associated antibiotic effects, especially the lack of resistance development, probably stem from the compound's polypharmacology.
RSC advancesQ3 · CHEMISTRY
Thompson loop: opportunities for antitubercular drug design by targeting the weak spot in demethylmenaquinone methyltransferase protein
Q3 · CHEMISTRY
ArticleOA
Author: Adewumi, Adeniyi T. ; Soliman, Mahmoud E. S. ; Ajadi, Mary B. ; Soremekun, Opeyemi S.
Graphical superimposed snapshots of the Thompson novel loop (yellow) of
Scientific reportsQ3 · CROSS-FIELD
Electricity from lignocellulosic substrates by thermophilic Geobacillus species
Q3 · CROSS-FIELD
ArticleOA
Author: Sani, Rajesh Kumar ; Gadhamshetty, Venkataramana ; Urgun-Demirtas, Meltem ; Shrestha, Namita ; Govil, Tanvi ; Tripathi, Abhilash Kumar ; Kasthuri, Venkateswaran
Abstract:
Given our vast lignocellulosic biomass reserves and the difficulty in bioprocessing them without expensive pretreatment and fuel separation steps, the conversion of lignocellulosic biomass directly into electricity would be beneficial. Here we report the previously unexplored capabilities of thermophilic Geobacillus sp. strain WSUCF1 to generate electricity directly from such complex substrates in microbial fuel cells. This process obviates the need for exogenous enzymes and redox mediator supplements. Cyclic voltammetry and chromatography studies revealed the electrochemical signatures of riboflavin molecules that reflect mediated electron transfer capabilities of strain WSUCF1. Proteomics and genomics analysis corroborated that WSUCF1 biofilms uses type-II NADH dehydrogenase and demethylmenaquinone methyltransferase to transfer the electrons to conducting anode via the redox active pheromone lipoproteins localized at the cell membrane.
Analysis
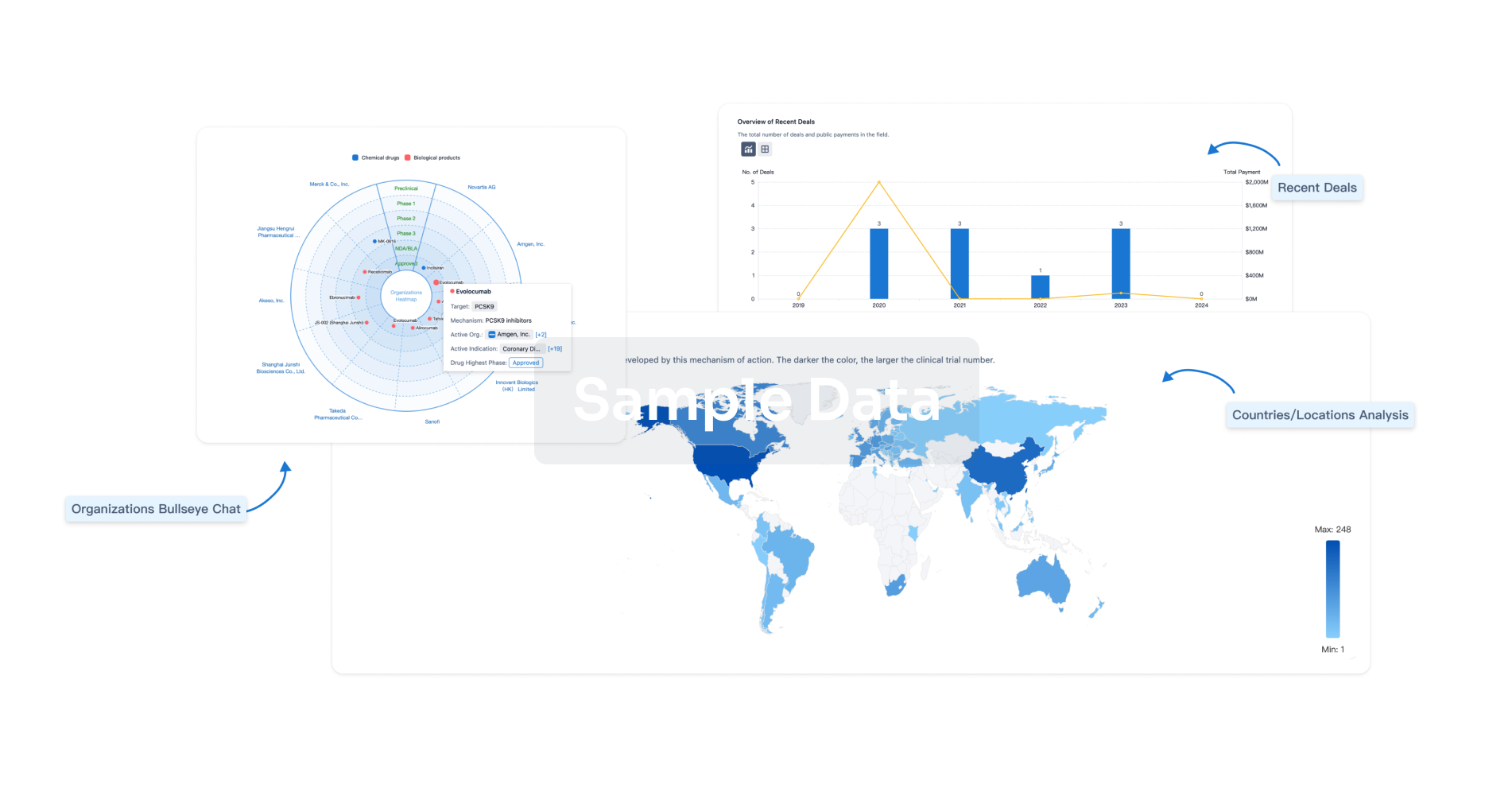
Perform a panoramic analysis of this field.
login
or
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.
Bio
Bio Sequences Search & Analysis
Sign up for free
Chemical
Chemical Structures Search & Analysis
Sign up for free