Request Demo
What is Agalsidase alfa used for?
14 June 2024
Agalsidase alfa is a recombinant form of the human enzyme alpha-galactosidase A, and it is primarily marketed under the trade name Replagal. This enzyme replacement therapy (ERT) is designed to treat patients with Fabry disease, a rare genetic disorder caused by a deficiency of the alpha-galactosidase A enzyme. Fabry disease leads to the accumulation of a specific type of fat, called globotriaosylceramide (GL-3 or Gb3), in various organs and tissues throughout the body, which can result in a wide range of symptoms and complications. Replagal has been developed by Shire, now part of Takeda Pharmaceutical Company, and it received approval in the European Union in 2001 and in several other countries subsequently. However, it has not been approved by the U.S. Food and Drug Administration (FDA).
Agalsidase alfa works by supplementing the deficient enzyme in patients with Fabry disease. The recombinant enzyme helps to break down the accumulated GL-3, thereby alleviating the symptoms and reducing the risk of long-term complications such as kidney failure, heart disease, and stroke. Clinical trials and various studies have shown that agalsidase alfa can effectively reduce GL-3 levels in organs and improve the quality of life for many individuals with Fabry disease. However, ongoing research and post-marketing surveillance continue to evaluate the long-term efficacy and safety of this treatment.
Agalsidase alfa exerts its therapeutic effect by specifically targeting and hydrolyzing the glycosphingolipid GL-3. In patients with Fabry disease, mutations in the GLA gene lead to a deficiency or complete lack of functional alpha-galactosidase A enzyme. This deficiency results in the progressive accumulation of GL-3 in lysosomes within cells, particularly in the vascular endothelium, heart, kidneys, and nervous system. By introducing recombinant alpha-galactosidase A into the patient’s system, agalsidase alfa facilitates the breakdown of GL-3 into simpler, non-toxic molecules that can be excreted by the body.
Once administered, agalsidase alfa is taken up by cells via receptor-mediated endocytosis, primarily through the mannose-6-phosphate receptor pathway. Inside the lysosomes, the enzyme catalyzes the hydrolysis of the terminal alpha-galactosyl moieties from GL-3, thus reducing its accumulation. This process helps to mitigate the pathogenic effects of GL-3 deposits and improve cellular function. As a result, patients can experience reductions in pain, gastrointestinal symptoms, and improved renal and cardiac function.
Agalsidase alfa is administered via intravenous infusion, typically in a hospital or clinical setting under the supervision of healthcare professionals. The recommended dosage for adults and children over seven years of age is 0.2 mg/kg body weight, administered once every two weeks. The infusion process usually takes about 40 minutes to an hour, although this can vary depending on the patient's specific circumstances and response to the treatment.
The onset of action for agalsidase alfa can vary among individuals. Some patients may experience symptomatic relief within a few weeks of starting therapy, while others may require several months to notice significant improvements. Regular infusions are essential to maintain enzyme levels and sustain therapeutic benefits. Periodic assessments, including clinical evaluations and laboratory tests, are recommended to monitor the effectiveness of the treatment and adjust the dosage if necessary.
As with any medication, agalsidase alfa can cause side effects, although not everyone will experience them. Some common side effects include infusion-related reactions such as chills, fever, headache, nausea, vomiting, and fatigue. These reactions are generally mild to moderate and can often be managed by slowing the infusion rate or pre-medicating with antihistamines and corticosteroids.
More severe side effects are less common but may include hypersensitivity reactions such as anaphylaxis, which requires immediate medical attention. Patients with a known hypersensitivity to agalsidase alfa or any of its excipients should not use this medication. Additionally, individuals with advanced renal disease may have a reduced response to enzyme replacement therapy, and the risks and benefits should be carefully weighed in such cases.
Other potential side effects include abdominal pain, diarrhea, upper respiratory tract infections, and increased levels of liver enzymes. Regular monitoring by healthcare providers is crucial to detect and manage any adverse effects promptly.
Certain medications may interact with agalsidase alfa, potentially affecting its efficacy or increasing the risk of side effects. Immunosuppressive drugs, such as cyclosporine and tacrolimus, may interfere with the uptake of agalsidase alfa by cells, reducing its therapeutic effect. Consequently, patients receiving immunosuppressive therapy should be closely monitored, and dosage adjustments may be necessary.
Additionally, other enzyme replacement therapies, such as agalsidase beta (Fabrazyme), may have overlapping effects with agalsidase alfa. While both medications aim to treat Fabry disease by providing the missing enzyme, they are not interchangeable and should not be used concurrently.
Patients should inform their healthcare providers about all the medications they are currently taking, including prescription drugs, over-the-counter medications, and dietary supplements. This information is crucial for ensuring that potential drug interactions are identified and managed appropriately.
In conclusion, agalsidase alfa represents a significant advancement in the treatment of Fabry disease, providing patients with a means to manage their condition and improve their quality of life. While the medication has proven effective in reducing GL-3 accumulation and alleviating symptoms, it is essential for patients to adhere to their treatment regimen and attend regular follow-up appointments to monitor their progress and address any potential side effects or drug interactions. As research continues, further insights into the long-term benefits and optimal use of agalsidase alfa are expected to emerge, offering hope to individuals affected by this rare and challenging disorder.
Agalsidase alfa works by supplementing the deficient enzyme in patients with Fabry disease. The recombinant enzyme helps to break down the accumulated GL-3, thereby alleviating the symptoms and reducing the risk of long-term complications such as kidney failure, heart disease, and stroke. Clinical trials and various studies have shown that agalsidase alfa can effectively reduce GL-3 levels in organs and improve the quality of life for many individuals with Fabry disease. However, ongoing research and post-marketing surveillance continue to evaluate the long-term efficacy and safety of this treatment.
Agalsidase alfa exerts its therapeutic effect by specifically targeting and hydrolyzing the glycosphingolipid GL-3. In patients with Fabry disease, mutations in the GLA gene lead to a deficiency or complete lack of functional alpha-galactosidase A enzyme. This deficiency results in the progressive accumulation of GL-3 in lysosomes within cells, particularly in the vascular endothelium, heart, kidneys, and nervous system. By introducing recombinant alpha-galactosidase A into the patient’s system, agalsidase alfa facilitates the breakdown of GL-3 into simpler, non-toxic molecules that can be excreted by the body.
Once administered, agalsidase alfa is taken up by cells via receptor-mediated endocytosis, primarily through the mannose-6-phosphate receptor pathway. Inside the lysosomes, the enzyme catalyzes the hydrolysis of the terminal alpha-galactosyl moieties from GL-3, thus reducing its accumulation. This process helps to mitigate the pathogenic effects of GL-3 deposits and improve cellular function. As a result, patients can experience reductions in pain, gastrointestinal symptoms, and improved renal and cardiac function.
Agalsidase alfa is administered via intravenous infusion, typically in a hospital or clinical setting under the supervision of healthcare professionals. The recommended dosage for adults and children over seven years of age is 0.2 mg/kg body weight, administered once every two weeks. The infusion process usually takes about 40 minutes to an hour, although this can vary depending on the patient's specific circumstances and response to the treatment.
The onset of action for agalsidase alfa can vary among individuals. Some patients may experience symptomatic relief within a few weeks of starting therapy, while others may require several months to notice significant improvements. Regular infusions are essential to maintain enzyme levels and sustain therapeutic benefits. Periodic assessments, including clinical evaluations and laboratory tests, are recommended to monitor the effectiveness of the treatment and adjust the dosage if necessary.
As with any medication, agalsidase alfa can cause side effects, although not everyone will experience them. Some common side effects include infusion-related reactions such as chills, fever, headache, nausea, vomiting, and fatigue. These reactions are generally mild to moderate and can often be managed by slowing the infusion rate or pre-medicating with antihistamines and corticosteroids.
More severe side effects are less common but may include hypersensitivity reactions such as anaphylaxis, which requires immediate medical attention. Patients with a known hypersensitivity to agalsidase alfa or any of its excipients should not use this medication. Additionally, individuals with advanced renal disease may have a reduced response to enzyme replacement therapy, and the risks and benefits should be carefully weighed in such cases.
Other potential side effects include abdominal pain, diarrhea, upper respiratory tract infections, and increased levels of liver enzymes. Regular monitoring by healthcare providers is crucial to detect and manage any adverse effects promptly.
Certain medications may interact with agalsidase alfa, potentially affecting its efficacy or increasing the risk of side effects. Immunosuppressive drugs, such as cyclosporine and tacrolimus, may interfere with the uptake of agalsidase alfa by cells, reducing its therapeutic effect. Consequently, patients receiving immunosuppressive therapy should be closely monitored, and dosage adjustments may be necessary.
Additionally, other enzyme replacement therapies, such as agalsidase beta (Fabrazyme), may have overlapping effects with agalsidase alfa. While both medications aim to treat Fabry disease by providing the missing enzyme, they are not interchangeable and should not be used concurrently.
Patients should inform their healthcare providers about all the medications they are currently taking, including prescription drugs, over-the-counter medications, and dietary supplements. This information is crucial for ensuring that potential drug interactions are identified and managed appropriately.
In conclusion, agalsidase alfa represents a significant advancement in the treatment of Fabry disease, providing patients with a means to manage their condition and improve their quality of life. While the medication has proven effective in reducing GL-3 accumulation and alleviating symptoms, it is essential for patients to adhere to their treatment regimen and attend regular follow-up appointments to monitor their progress and address any potential side effects or drug interactions. As research continues, further insights into the long-term benefits and optimal use of agalsidase alfa are expected to emerge, offering hope to individuals affected by this rare and challenging disorder.
How to obtain the latest development progress of all drugs?
In the Synapse database, you can stay updated on the latest research and development advances of all drugs. This service is accessible anytime and anywhere, with updates available daily or weekly. Use the "Set Alert" function to stay informed. Click on the image below to embark on a brand new journey of drug discovery!
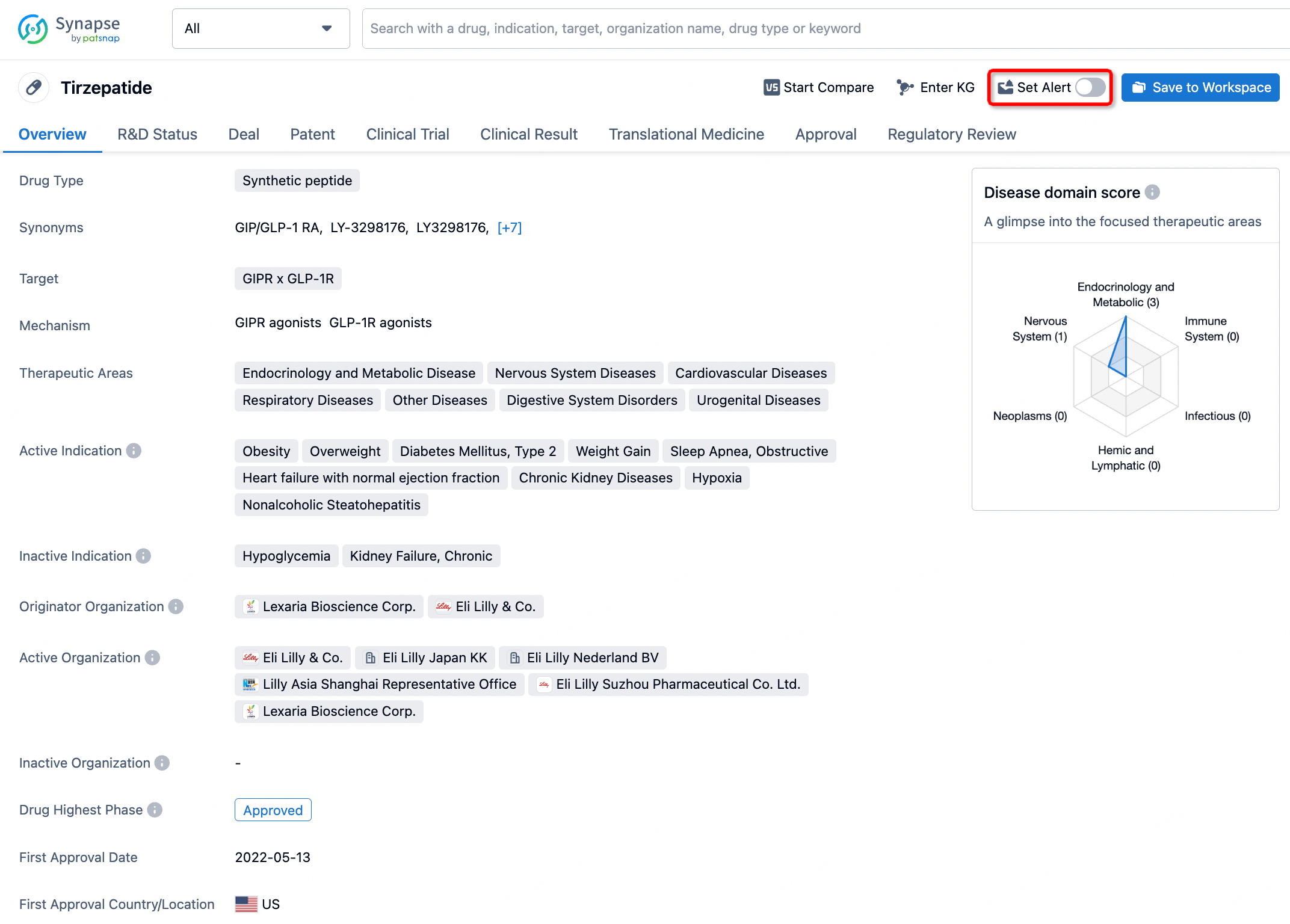
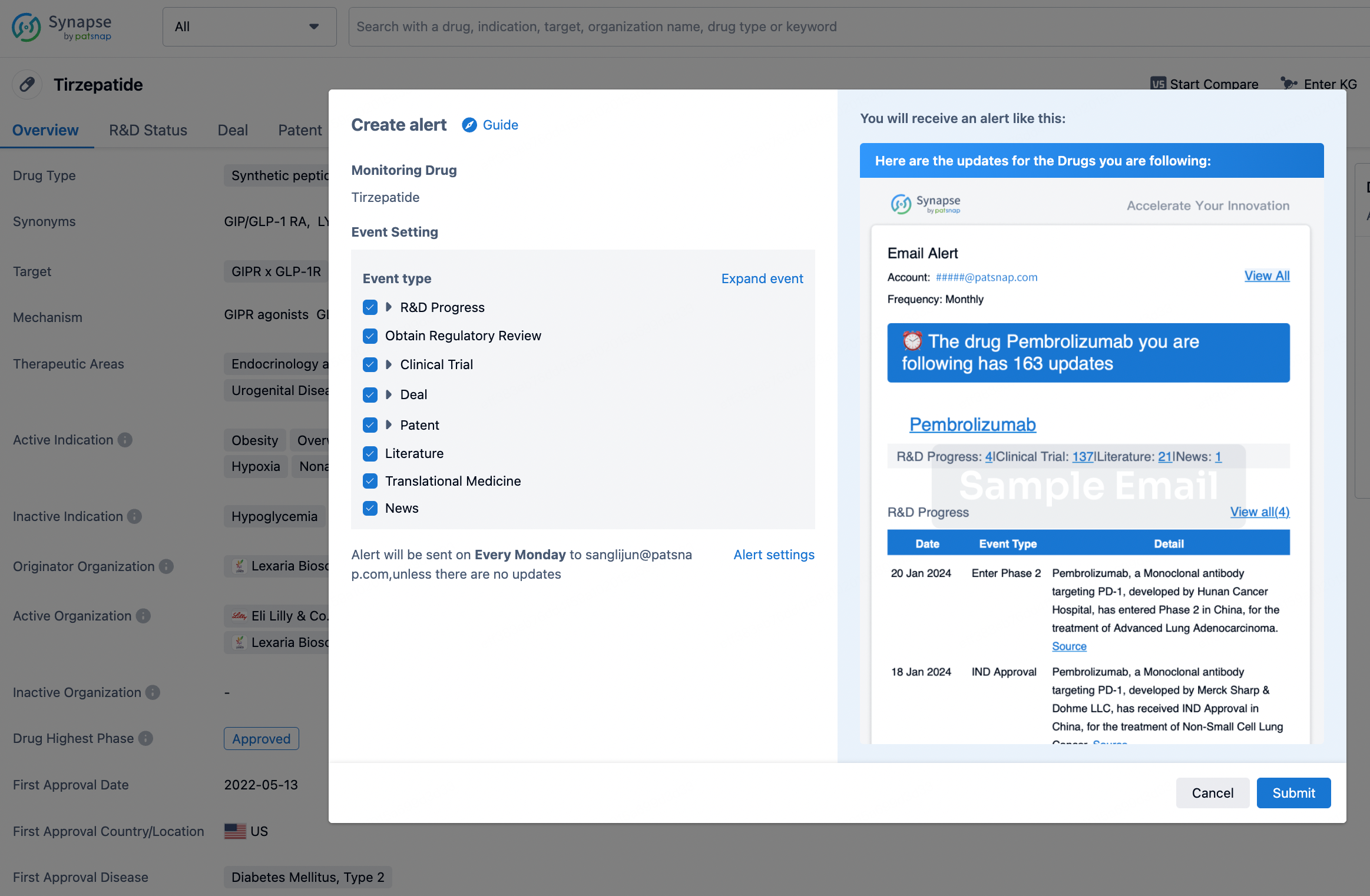
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.