Request Demo
What is Ibiglustat used for?
28 June 2024
Ibiglustat is an investigational drug that has generated significant interest in the medical and scientific communities due to its potential therapeutic benefits. Developed by various leading research institutions, Ibiglustat is a small-molecule inhibitor that primarily targets the enzyme glucosylceramide synthase (GCS). By inhibiting this enzyme, Ibiglustat aims to modulate glycosphingolipid metabolism, which has shown promise in treating a variety of lysosomal storage disorders and other metabolic conditions. As of the latest research updates, Ibiglustat is still undergoing clinical trials to ascertain its efficacy and safety profiles, but early results have been promising.
In terms of drug type, Ibiglustat is classified as a small-molecule, orally available inhibitor. The oral bioavailability of this compound makes it particularly attractive for long-term treatments, as it improves patient compliance compared to intravenous therapies. The main indications for Ibiglustat include Gaucher disease and Fabry disease, both of which are rare genetic disorders characterized by dysfunctional metabolism of glycosphingolipids. Research progress has seen the transition of Ibiglustat from preclinical studies to phase 2 and 3 clinical trials, where its effectiveness and safety are being evaluated in human subjects.
The mechanism of action of Ibiglustat is centered around its ability to inhibit glucosylceramide synthase. This enzyme is pivotal in the biosynthetic pathway of glycosphingolipids, a complex class of lipids involved in various cellular processes, including cell signaling and membrane structure. In conditions like Gaucher and Fabry diseases, mutations lead to a deficiency of specific lysosomal enzymes responsible for breaking down glycosphingolipids. This deficiency results in the accumulation of these lipids within cells, causing a myriad of symptoms and complications.
Ibiglustat intervenes in this pathological process by preventing the initial step in glycosphingolipid synthesis. By inhibiting glucosylceramide synthase, the drug effectively reduces the production of glucosylceramide and subsequent glycosphingolipids, thereby alleviating the burden on lysosomal degradation pathways. This reduction in substrate availability helps to ameliorate the clinical manifestations of lysosomal storage disorders. It is worth noting that the selectivity and potency of Ibiglustat in targeting GCS are critical factors that contribute to its therapeutic potential.
The primary indication of Ibiglustat is Gaucher disease, which is a hereditary lysosomal storage disorder caused by a deficiency in the enzyme glucocerebrosidase. This leads to the accumulation of glucocerebroside in various organs, including the spleen, liver, and bone marrow. Patients with Gaucher disease often experience symptoms such as anemia, thrombocytopenia, bone pain, and organomegaly. Traditional treatments include enzyme replacement therapy (ERT) and substrate reduction therapy (SRT). However, these treatments have limitations, including the need for frequent infusions and potential side effects.
Ibiglustat offers a novel approach by directly inhibiting the synthesis of the substrate that accumulates in Gaucher disease. This could potentially reduce the frequency of treatments and improve the quality of life for patients. Clinical trials are ongoing to determine the efficacy of Ibiglustat in reducing disease symptoms and improving biomarkers in patients with Gaucher disease. Preliminary results have shown promise, indicating significant reductions in glucocerebroside levels and improvements in clinical outcomes.
Additionally, Ibiglustat is being investigated for its potential in treating Fabry disease, another lysosomal storage disorder caused by a deficiency in the enzyme alpha-galactosidase A. This deficiency results in the accumulation of globotriaosylceramide (Gb3) in various tissues, leading to symptoms such as pain, kidney dysfunction, heart disease, and stroke. Current treatments for Fabry disease include ERT and SRT, but these therapies also have limitations.
Ibiglustat's mechanism of action could similarly benefit Fabry disease patients by reducing the synthesis of glycosphingolipids and thus decreasing the accumulation of harmful substrates. Ongoing research aims to validate this hypothesis and expand the therapeutic indications of Ibiglustat.
In conclusion, Ibiglustat represents a promising therapeutic candidate for the treatment of lysosomal storage disorders like Gaucher and Fabry diseases. By targeting the root cause of these conditions through the inhibition of glucosylceramide synthase, Ibiglustat has the potential to offer a more effective and convenient treatment option for patients. As clinical trials continue to unfold, the medical community eagerly awaits more comprehensive data to fully understand the benefits and limitations of this innovative drug.
In terms of drug type, Ibiglustat is classified as a small-molecule, orally available inhibitor. The oral bioavailability of this compound makes it particularly attractive for long-term treatments, as it improves patient compliance compared to intravenous therapies. The main indications for Ibiglustat include Gaucher disease and Fabry disease, both of which are rare genetic disorders characterized by dysfunctional metabolism of glycosphingolipids. Research progress has seen the transition of Ibiglustat from preclinical studies to phase 2 and 3 clinical trials, where its effectiveness and safety are being evaluated in human subjects.
The mechanism of action of Ibiglustat is centered around its ability to inhibit glucosylceramide synthase. This enzyme is pivotal in the biosynthetic pathway of glycosphingolipids, a complex class of lipids involved in various cellular processes, including cell signaling and membrane structure. In conditions like Gaucher and Fabry diseases, mutations lead to a deficiency of specific lysosomal enzymes responsible for breaking down glycosphingolipids. This deficiency results in the accumulation of these lipids within cells, causing a myriad of symptoms and complications.
Ibiglustat intervenes in this pathological process by preventing the initial step in glycosphingolipid synthesis. By inhibiting glucosylceramide synthase, the drug effectively reduces the production of glucosylceramide and subsequent glycosphingolipids, thereby alleviating the burden on lysosomal degradation pathways. This reduction in substrate availability helps to ameliorate the clinical manifestations of lysosomal storage disorders. It is worth noting that the selectivity and potency of Ibiglustat in targeting GCS are critical factors that contribute to its therapeutic potential.
The primary indication of Ibiglustat is Gaucher disease, which is a hereditary lysosomal storage disorder caused by a deficiency in the enzyme glucocerebrosidase. This leads to the accumulation of glucocerebroside in various organs, including the spleen, liver, and bone marrow. Patients with Gaucher disease often experience symptoms such as anemia, thrombocytopenia, bone pain, and organomegaly. Traditional treatments include enzyme replacement therapy (ERT) and substrate reduction therapy (SRT). However, these treatments have limitations, including the need for frequent infusions and potential side effects.
Ibiglustat offers a novel approach by directly inhibiting the synthesis of the substrate that accumulates in Gaucher disease. This could potentially reduce the frequency of treatments and improve the quality of life for patients. Clinical trials are ongoing to determine the efficacy of Ibiglustat in reducing disease symptoms and improving biomarkers in patients with Gaucher disease. Preliminary results have shown promise, indicating significant reductions in glucocerebroside levels and improvements in clinical outcomes.
Additionally, Ibiglustat is being investigated for its potential in treating Fabry disease, another lysosomal storage disorder caused by a deficiency in the enzyme alpha-galactosidase A. This deficiency results in the accumulation of globotriaosylceramide (Gb3) in various tissues, leading to symptoms such as pain, kidney dysfunction, heart disease, and stroke. Current treatments for Fabry disease include ERT and SRT, but these therapies also have limitations.
Ibiglustat's mechanism of action could similarly benefit Fabry disease patients by reducing the synthesis of glycosphingolipids and thus decreasing the accumulation of harmful substrates. Ongoing research aims to validate this hypothesis and expand the therapeutic indications of Ibiglustat.
In conclusion, Ibiglustat represents a promising therapeutic candidate for the treatment of lysosomal storage disorders like Gaucher and Fabry diseases. By targeting the root cause of these conditions through the inhibition of glucosylceramide synthase, Ibiglustat has the potential to offer a more effective and convenient treatment option for patients. As clinical trials continue to unfold, the medical community eagerly awaits more comprehensive data to fully understand the benefits and limitations of this innovative drug.
How to obtain the latest development progress of all drugs?
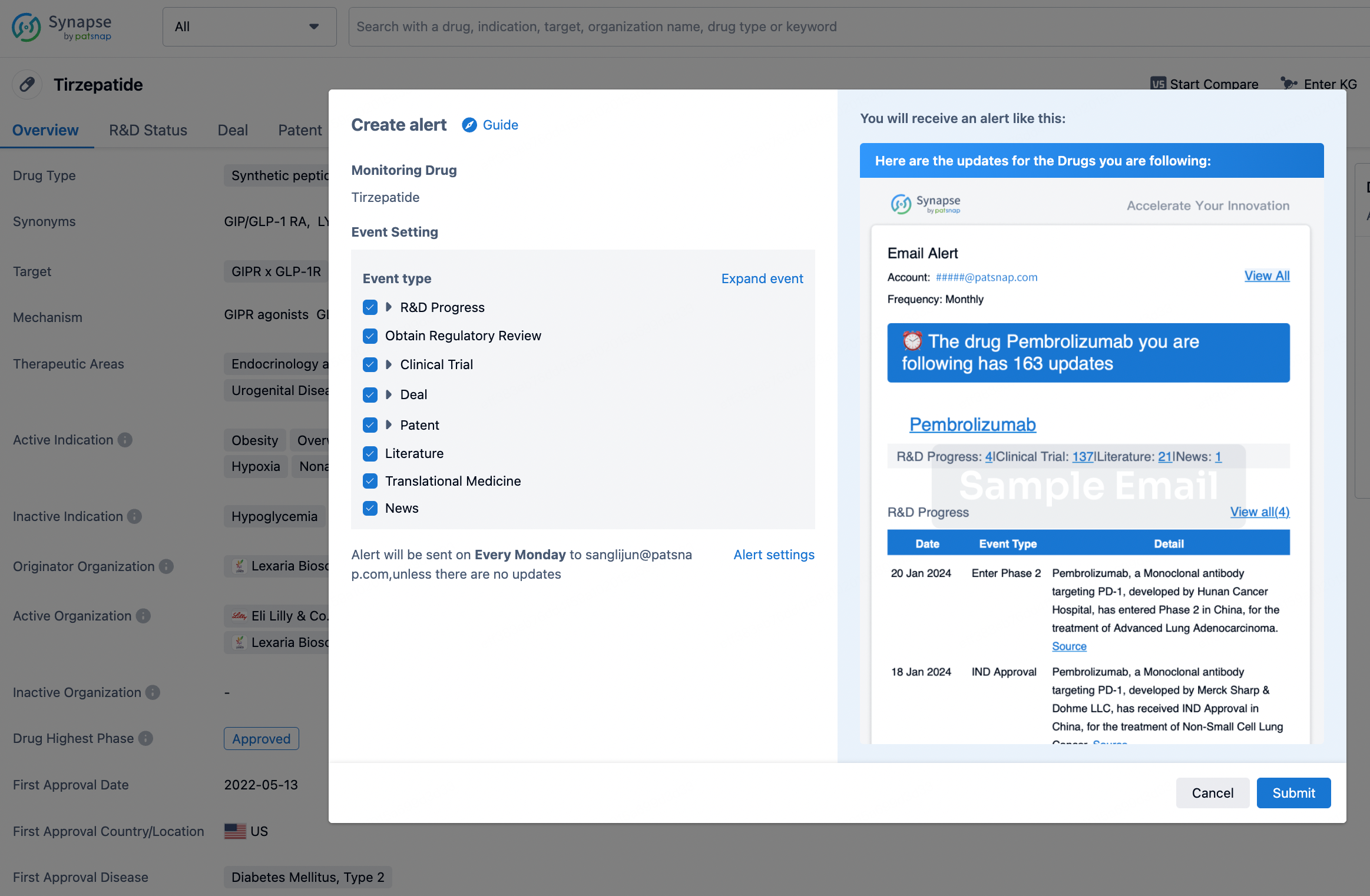
In the Synapse database, you can stay updated on the latest research and development advances of all drugs. This service is accessible anytime and anywhere, with updates available daily or weekly. Use the "Set Alert" function to stay informed. Click on the image below to embark on a brand new journey of drug discovery!
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.