Request Demo
What is the mechanism of Golodirsen?
17 July 2024
Golodirsen is a medication developed as a treatment for Duchenne muscular dystrophy (DMD), a severe type of muscular dystrophy caused by mutations in the dystrophin gene. This gene is crucial for the production of dystrophin, a protein that helps stabilize and protect muscle fibers. In patients with DMD, the absence or deficiency of functional dystrophin leads to progressive muscle degeneration and weakness. Golodirsen is designed to address this underlying cause through a mechanism known as exon skipping.
Golodirsen specifically targets a region of the dystrophin gene called exon 53. Mutations in the dystrophin gene often result in premature stop codons that truncate the protein, rendering it nonfunctional. By inducing exon 53 skipping during mRNA processing, Golodirsen aims to restore the reading frame of the dystrophin mRNA, allowing the production of a shorter but still functional dystrophin protein. This therapeutic approach relies on antisense oligonucleotides (AONs), which are short DNA or RNA molecules that can bind to specific sequences of RNA.
Here is a detailed look at how Golodirsen works:
1. **Binding to Pre-mRNA**: Golodirsen is an antisense oligonucleotide that binds to the pre-mRNA of the dystrophin gene. It is designed to specifically attach to the sequences around exon 53.
2. **Exon Skipping**: Once bound, Golodirsen alters the splicing of the pre-mRNA. Normally, the cellular machinery would include exon 53 in the final mRNA, but Golodirsen causes this exon to be skipped. By doing so, it helps to bypass the mutation that causes the defective dystrophin protein.
3. **Restoration of Reading Frame**: Skipping exon 53 allows the remaining exons to be joined together in a way that restores the reading frame of the mRNA. This corrected mRNA can then be translated into a shorter but functional dystrophin protein.
4. **Protein Production**: The resulting dystrophin protein, although not entirely identical to the full-length version, retains much of its functionality. This partially functional dystrophin can help to stabilize muscle cell membranes, thus slowing the progression of muscle degeneration in patients with DMD.
5. **Clinical Outcome**: The production of a near-functional dystrophin protein can lead to improved muscle function and a slower decline in muscle strength and mobility. This can significantly enhance the quality of life for individuals suffering from DMD.
The effectiveness of Golodirsen and other exon-skipping therapies is still an area of active research. Clinical trials are conducted to determine the long-term benefits and potential side effects of these treatments. However, early results have shown promise, with some patients experiencing increased dystrophin levels and improved muscle function.
In summary, Golodirsen operates through a sophisticated mechanism of exon skipping to correct the genetic defect in the dystrophin gene that causes Duchenne muscular dystrophy. By promoting the production of a functional dystrophin protein, it offers hope for slowing down the progression of this debilitating disease.
Golodirsen specifically targets a region of the dystrophin gene called exon 53. Mutations in the dystrophin gene often result in premature stop codons that truncate the protein, rendering it nonfunctional. By inducing exon 53 skipping during mRNA processing, Golodirsen aims to restore the reading frame of the dystrophin mRNA, allowing the production of a shorter but still functional dystrophin protein. This therapeutic approach relies on antisense oligonucleotides (AONs), which are short DNA or RNA molecules that can bind to specific sequences of RNA.
Here is a detailed look at how Golodirsen works:
1. **Binding to Pre-mRNA**: Golodirsen is an antisense oligonucleotide that binds to the pre-mRNA of the dystrophin gene. It is designed to specifically attach to the sequences around exon 53.
2. **Exon Skipping**: Once bound, Golodirsen alters the splicing of the pre-mRNA. Normally, the cellular machinery would include exon 53 in the final mRNA, but Golodirsen causes this exon to be skipped. By doing so, it helps to bypass the mutation that causes the defective dystrophin protein.
3. **Restoration of Reading Frame**: Skipping exon 53 allows the remaining exons to be joined together in a way that restores the reading frame of the mRNA. This corrected mRNA can then be translated into a shorter but functional dystrophin protein.
4. **Protein Production**: The resulting dystrophin protein, although not entirely identical to the full-length version, retains much of its functionality. This partially functional dystrophin can help to stabilize muscle cell membranes, thus slowing the progression of muscle degeneration in patients with DMD.
5. **Clinical Outcome**: The production of a near-functional dystrophin protein can lead to improved muscle function and a slower decline in muscle strength and mobility. This can significantly enhance the quality of life for individuals suffering from DMD.
The effectiveness of Golodirsen and other exon-skipping therapies is still an area of active research. Clinical trials are conducted to determine the long-term benefits and potential side effects of these treatments. However, early results have shown promise, with some patients experiencing increased dystrophin levels and improved muscle function.
In summary, Golodirsen operates through a sophisticated mechanism of exon skipping to correct the genetic defect in the dystrophin gene that causes Duchenne muscular dystrophy. By promoting the production of a functional dystrophin protein, it offers hope for slowing down the progression of this debilitating disease.
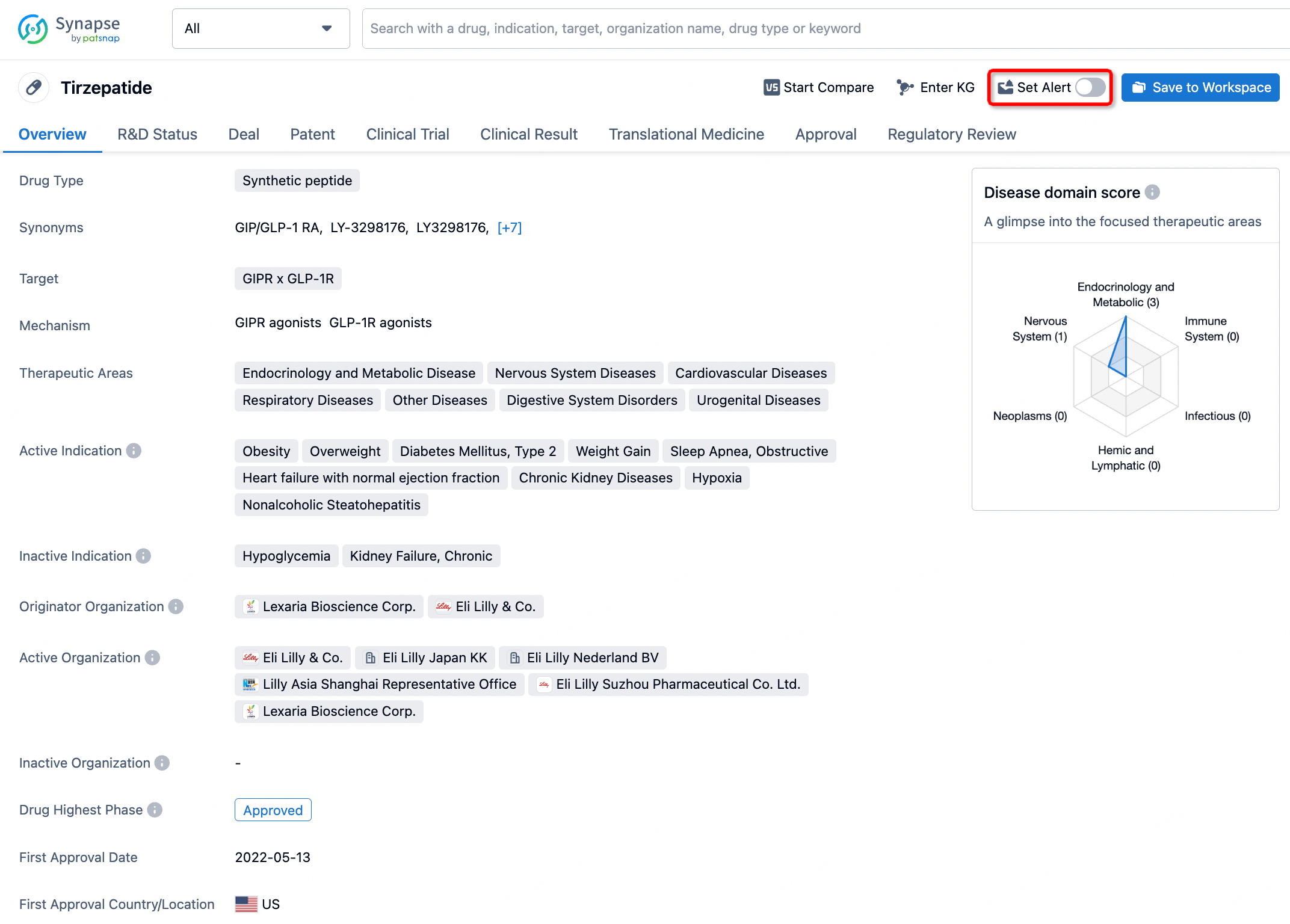
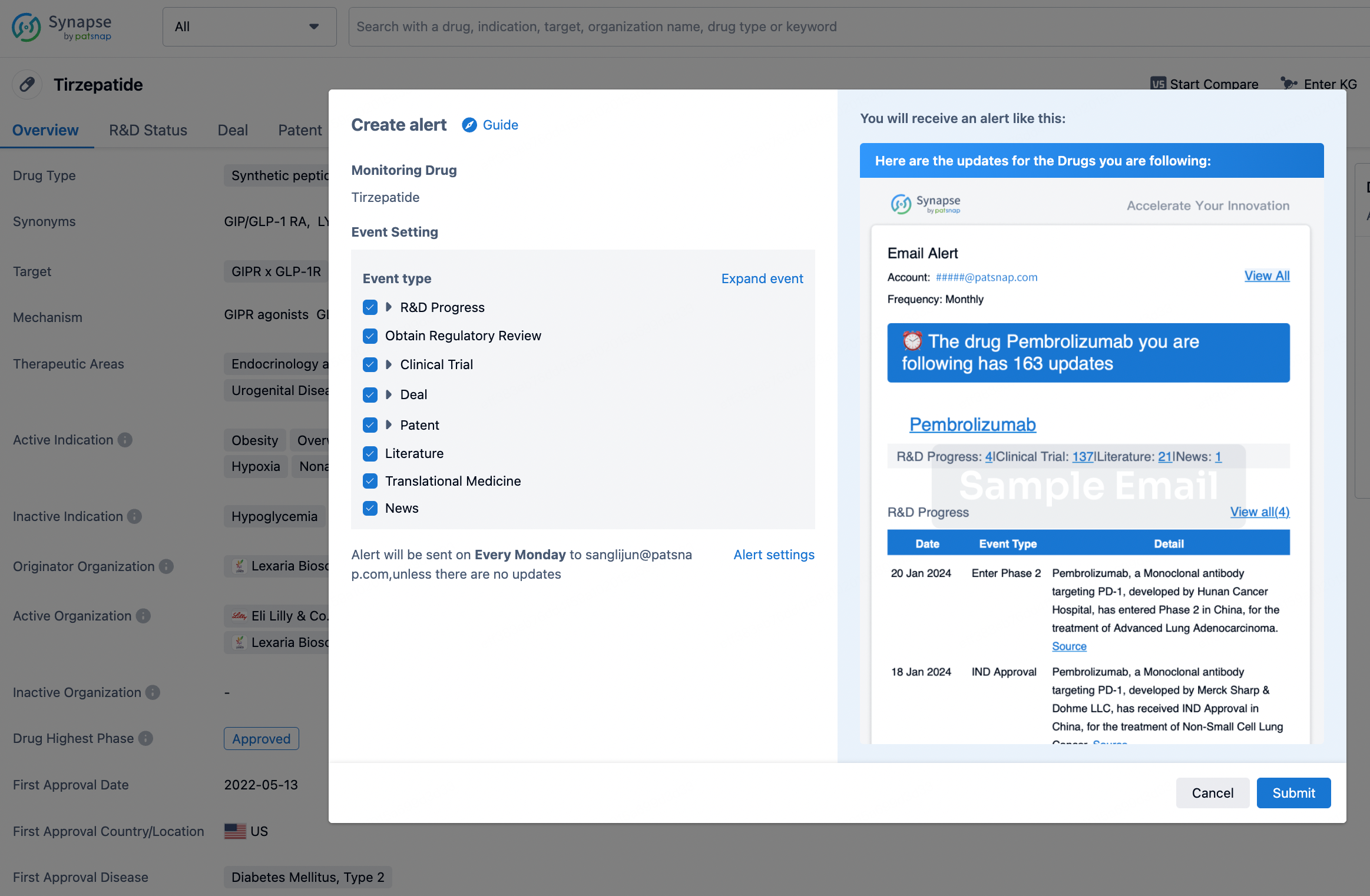
How to obtain the latest development progress of all drugs?
In the Synapse database, you can stay updated on the latest research and development advances of all drugs. This service is accessible anytime and anywhere, with updates available daily or weekly. Use the "Set Alert" function to stay informed. Click on the image below to embark on a brand new journey of drug discovery!
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.