Request Demo
What is the mechanism of Ivacaftor?
17 July 2024
Ivacaftor is a groundbreaking pharmaceutical agent that has significantly impacted the treatment of cystic fibrosis (CF), a genetic disorder that primarily affects the lungs and digestive system. To understand its mechanism, it is essential to delve into the underlying genetic and cellular abnormalities that cause cystic fibrosis and how Ivacaftor interacts with these abnormalities to bring about therapeutic benefits.
Cystic fibrosis is caused by mutations in the CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) gene. The CFTR protein produced by this gene functions as a chloride channel, which is crucial for the regulation of salt and water transport across cell membranes. In individuals with cystic fibrosis, mutations in the CFTR gene lead to defective or deficient CFTR protein, resulting in impaired chloride transport. This impairment causes the production of thick, sticky mucus that can obstruct airways, promote bacterial infections, and lead to progressive lung damage.
The most common mutation seen in cystic fibrosis patients is the F508del mutation, but Ivacaftor specifically targets a different subset of CFTR mutations, particularly gating mutations such as G551D. Gating mutations affect the protein's ability to open properly, thereby reducing chloride ion flow through the CFTR channel.
Ivacaftor works by directly binding to the defective CFTR protein produced by these gating mutations, enhancing its function. It acts as a CFTR potentiator, meaning it increases the probability that the CFTR channels will remain open. This allows for a more efficient chloride ion transport across the cell membrane, which helps to restore the balance of salt and water on the epithelial surfaces, thus improving hydration and thinning of the mucus. Consequently, this alleviates many of the symptoms associated with cystic fibrosis, such as chronic cough, recurrent lung infections, and impaired lung function.
The efficacy of Ivacaftor has been demonstrated in multiple clinical trials. Patients with specific CFTR gating mutations treated with Ivacaftor showed significant improvements in lung function (measured by forced expiratory volume in one second, FEV1), weight gain, and reduction in pulmonary exacerbations. These clinical benefits are attributed directly to the improved function of the CFTR protein.
It is important to note that Ivacaftor is not a cure for cystic fibrosis, but it represents a substantial advancement in the treatment paradigm for patients with specific CFTR mutations. By targeting the underlying defect at a molecular level, Ivacaftor offers a more precise and effective treatment option compared to traditional therapies that primarily address symptoms rather than the root cause.
In summary, the mechanism of Ivacaftor involves its role as a CFTR potentiator, where it enhances the gating function of the CFTR protein in patients with specific mutations. By improving chloride ion transport, Ivacaftor alleviates the characteristic symptoms of cystic fibrosis, offering significant clinical benefits and improving the quality of life for affected individuals.
Cystic fibrosis is caused by mutations in the CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) gene. The CFTR protein produced by this gene functions as a chloride channel, which is crucial for the regulation of salt and water transport across cell membranes. In individuals with cystic fibrosis, mutations in the CFTR gene lead to defective or deficient CFTR protein, resulting in impaired chloride transport. This impairment causes the production of thick, sticky mucus that can obstruct airways, promote bacterial infections, and lead to progressive lung damage.
The most common mutation seen in cystic fibrosis patients is the F508del mutation, but Ivacaftor specifically targets a different subset of CFTR mutations, particularly gating mutations such as G551D. Gating mutations affect the protein's ability to open properly, thereby reducing chloride ion flow through the CFTR channel.
Ivacaftor works by directly binding to the defective CFTR protein produced by these gating mutations, enhancing its function. It acts as a CFTR potentiator, meaning it increases the probability that the CFTR channels will remain open. This allows for a more efficient chloride ion transport across the cell membrane, which helps to restore the balance of salt and water on the epithelial surfaces, thus improving hydration and thinning of the mucus. Consequently, this alleviates many of the symptoms associated with cystic fibrosis, such as chronic cough, recurrent lung infections, and impaired lung function.
The efficacy of Ivacaftor has been demonstrated in multiple clinical trials. Patients with specific CFTR gating mutations treated with Ivacaftor showed significant improvements in lung function (measured by forced expiratory volume in one second, FEV1), weight gain, and reduction in pulmonary exacerbations. These clinical benefits are attributed directly to the improved function of the CFTR protein.
It is important to note that Ivacaftor is not a cure for cystic fibrosis, but it represents a substantial advancement in the treatment paradigm for patients with specific CFTR mutations. By targeting the underlying defect at a molecular level, Ivacaftor offers a more precise and effective treatment option compared to traditional therapies that primarily address symptoms rather than the root cause.
In summary, the mechanism of Ivacaftor involves its role as a CFTR potentiator, where it enhances the gating function of the CFTR protein in patients with specific mutations. By improving chloride ion transport, Ivacaftor alleviates the characteristic symptoms of cystic fibrosis, offering significant clinical benefits and improving the quality of life for affected individuals.
How to obtain the latest development progress of all drugs?
In the Synapse database, you can stay updated on the latest research and development advances of all drugs. This service is accessible anytime and anywhere, with updates available daily or weekly. Use the "Set Alert" function to stay informed. Click on the image below to embark on a brand new journey of drug discovery!
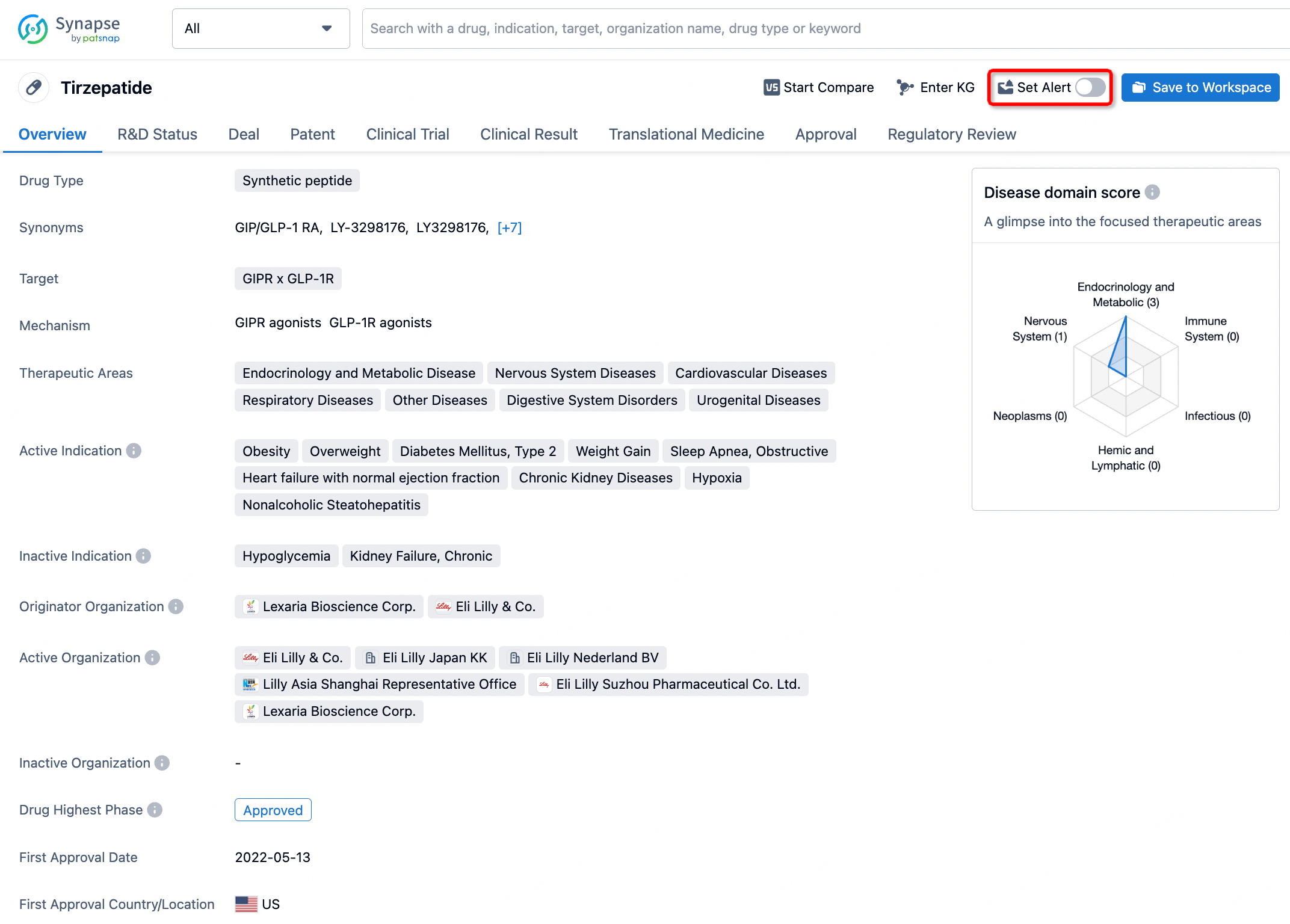
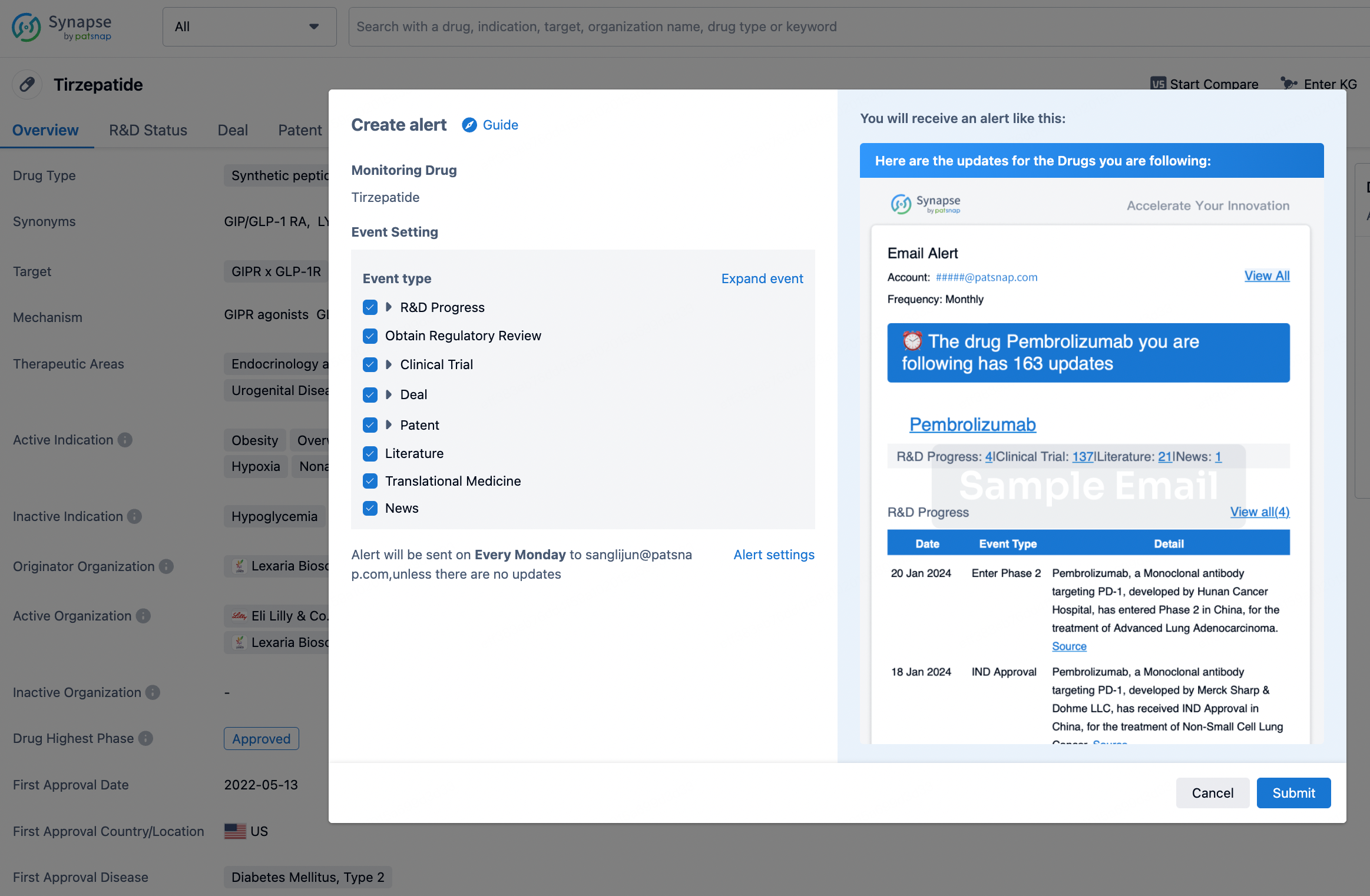
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.