Iptacopan - Novartis' first-in-class oral medication for PNH
Iptacopan, also known as LNP023, is the first oral small moleculecompletion factor B (CFB) inhibitor developed by Novartis Pharma AG. On June 5, 2023, for paroxysmal nocturnal hemoglobinuria (PNH), NMPA included Iptacopan in the priority review procedure.
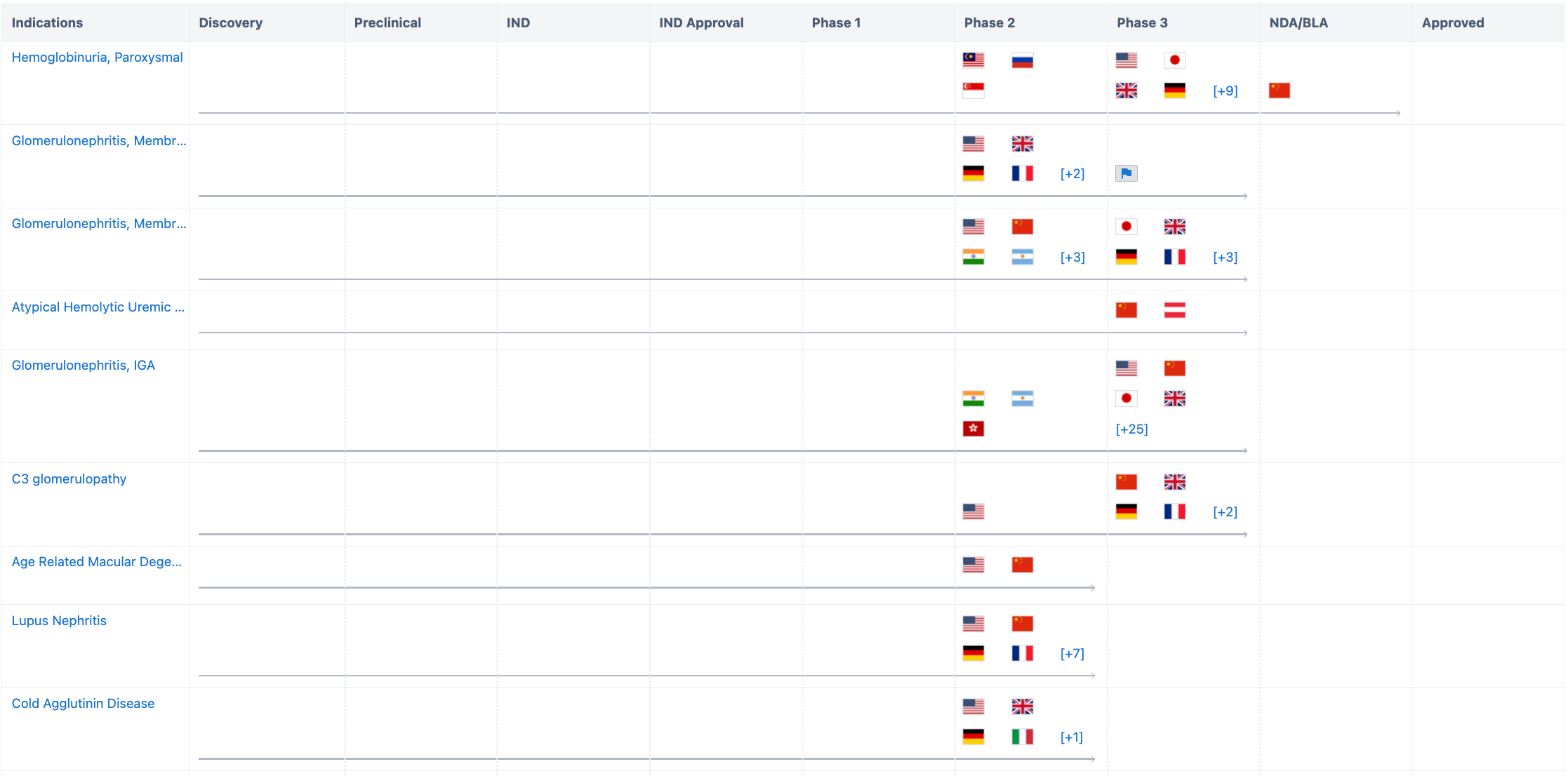
According to “Synapse”, Iptacopan has more than a dozen indications under study, and all the indications, such as Hemoglobinuria, Paroxysmal; Glomerulonephritis, Membranoproliferative; Glomerulonephritis, Membranous; Atypical Hemolytic Uremic Syndrome; Glomerulonephritis, IGA; C3 glomerulopathy; Age Related Macular Degeneration; Lupus Nephritis; Cold Agglutinin Disease; Purpura, Thrombocytopenic, Idiopathic, have entered the clinical trial stage. For more detailed information on R&D status, core patents, analysis, etc., please click on the image link below.
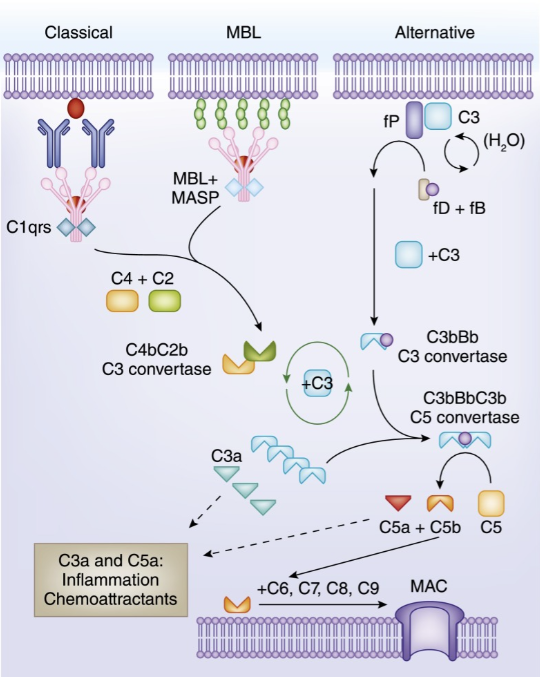
Mechanism of Action
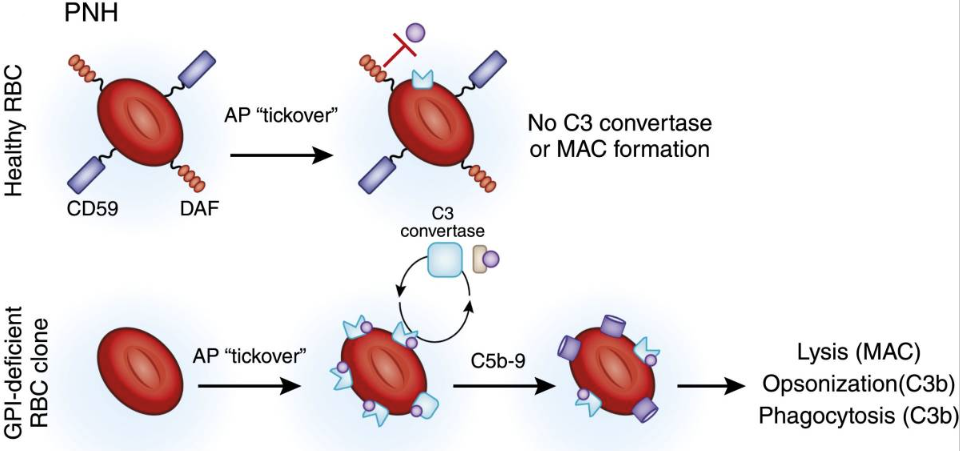
PNH is a rare hematological disorder affecting ∼1 to 1.5 per million individuals worldwide, caused by somatic mutations in the PIGA (phosphatidylinositol glycan A) gene in hematopoietic stem cells (HSCs).1,2 The PIGA mutations lead to a deficiency of glycosylphosphatidylinositol-anchored proteins, resulting in ineffective inhibition of the complement alternative pathway with increased susceptibility of PNH-type erythrocytes to intravascular hemolysis, spontaneous RBC lysis (membrane attack complex [MAC]) and leading to hemolytic anemia, impaired bone marrow (BM) function, and potentially life-threatening thrombotic events. CFB is a key component of the complement alternative pathway. As a CFB inhibitor, Iptacopan can inhibit the Complement system by blocking the binding of CFB and its receptor, thereby regulating and inhibiting inflammatory and immune responses, and alleviating tissue damage and disease pathological processes caused by inflammation.
Clinical trial
In an ongoing open-label phase 2 study (NCT03896152), PNH patients with active hemolysis were randomized to receive single-agent iptacopan twice daily at a dose of either 25 mg for 4 weeks followed by 100 mg for up to 2 years (cohort 1) or 50 mg for 4 weeks followed by 200 mg for up to 2 years (cohort 2). At the time of interim analysis, of 13 PNH patients enrolled, all 12 evaluable for efficacy achieved the primary endpoint of reduction in serum lactate dehydrogenase (LDH) levels by ≥60% by week 12 compared with baseline; mean LDH levels dropped rapidly and durably, namely by 77% and 85% at week 2 and by 86% and 86% at week 12 in cohorts 1 and 2, respectively. Most patients achieved a clinically meaningful improvement in hemoglobin (Hb) levels, and all but 1 patient remained transfusion-free up to week 12. Other markers of hemolysis, including bilirubin, reticulocytes, and haptoglobin, showed consistent improvements. Safety: No thromboembolic events were reported, and iptacopan was well tolerated, with no severe or serious adverse events reported.
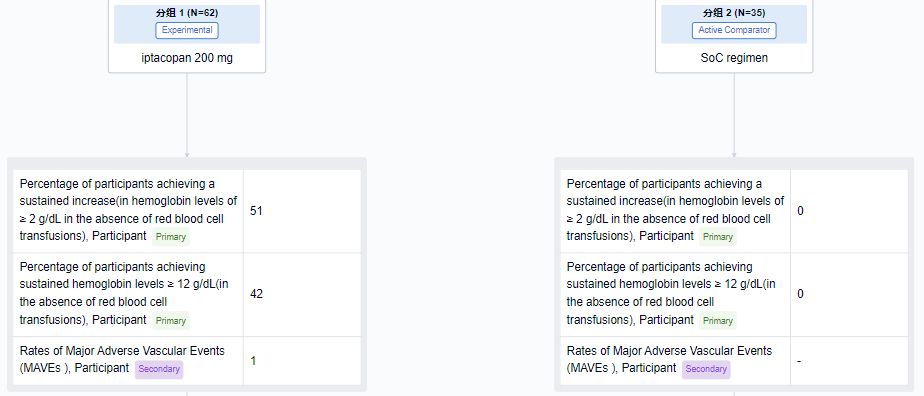
In a phase 3 clinical study on PNH (NCT04558918), the primary endpoint was the percentage of subjects whose Hb levels continued to increase by ≥ 2 g/dL from baseline without red blood cell cell infusion. The results showed that the proportion of Iptacopan group was 51%, while the standard treatment (Eculizumab or Ravulizumab) group was 0; The proportion of Iptacopan group with a continuous increase of 12 g/dL from baseline was 42%, while the standard treatment group was 0. Compared with standard treatment, Iptacopan showed superior efficacy.
Competitive Landscape
The current standard of care for PNH consists of monotherapy with either eculizumab or ravulizumab, both anticomplement component 5 (C5) monoclonal antibodies that significantly improve the clinical course of PNH patients. Nevertheless, a significant proportion of patients remain anemic and continue to require transfusions, largely due to persistent, C3-mediated extravascular hemolysis. Iptacopan targets the C3 pathway, and its emergence can to some extent compensate for this unmet clinical need.