Nature: Design of a Novel Bivalent Analgesic Targeting the μ-Opioid Receptor
Opioid receptors play a pivotal role in analgesic therapy, where their activation can inhibit the transmission of pain signals, significantly alleviating various types of acute and chronic pain, such as postoperative pain and cancer pain. Specific agonists for μ, δ, κ receptor subtypes, such as morphine, hydrocodone, and fentanyl, bind selectively to their corresponding receptors, triggering intracellular signaling pathways, and decreasing neuronal excitability, thus producing potent analgesic effects. However, opioid receptor-mediated analgesia also comes with a series of challenges. Long-term use can lead to tolerance, necessitating escalating doses to maintain efficacy, and may cause physiological and psychological dependence, increasing the risk of addiction. Some opioid analgesics have a range of adverse reactions, such as respiratory depression, constipation, nausea, and vomiting, which can be life-threatening in severe cases. Withdrawal symptoms may appear after discontinuation, and a gradual dose reduction or adjunct medication may be required.
Taking fentanyl as an example, it has been widely used as an opioid analgesic for anesthesia, breakthrough cancer pain or round-the-clock pain management. As a potent opioid receptor agonist, fentanyl was developed in the 1950s to meet the demand for strong and rapid analgesia, and it is closely related to other opioids such as morphine and oxycodone. Fentanyl's high potency has also made it a common adulterant in illicit drugs, particularly in heroin. According to reports, in 2017 alone, the United States witnessed 47,600 cases of drug overdose deaths involving some form of opioid (constituting over two-thirds of all overdose deaths).
The classical doctrine suggests that μOR-mediated analgesia, respiratory depression, and physical dependence are dependent on the downstream signaling pathways of different G proteins and β-arrestin-2. By designing functionally selective drugs, there is the potential to minimize the clinical side effects of opioids to the greatest extent possible. μOR is coupled with six different isoforms of G proteins, and the possibility of dissociating analgesic effects from adverse reactions by selectively targeting some of these isoforms remains largely unexplored.
A study published in Nature in 2023, titled "Structure-based design of bitopic ligands for the µ-opioid receptor," revealed a structure-guided strategy for designing bitopic ligands aimed at developing a new generation of analgesic drug candidates targeting the µ-opioid receptor. Researchers innovatively utilized dual-site ligand structures that bind simultaneously to the main active site and an auxiliary site of the receptor, aiming to precisely modulate opioid receptor signaling and effectively dissociate analgesic efficacy from side effects.

The researchers used molecular docking and dynamics simulations to guide the design of the novel bitopic ligands, starting with fentanyl as the compound of reference, and focusing on the highly conserved and functionally crucial sodium ion binding pocket within the receptor, which is considered an ideal allosteric site for developing unique opioid receptor modulators. By meticulously synthesizing and characterizing new compounds, and resolving the high-resolution structure of the receptor-ligand complexes using cryo-electron microscopy, the research team elucidated the intricate interactions between the bitopic ligands and the µOR. Experimental evidence demonstrated that these designed bitopic ligands exhibit superior analgesic effects while significantly reducing typical side effects such as respiratory depression.
Animal model studies further confirmed their in vivo stability and pharmacokinetic properties, suggesting good translational potential. The researchers also discovered that these ligands could selectively activate specific G protein subtypes, laying the theoretical groundwork for designing future opioid drugs with improved functional selectivity. As understanding deepens regarding the structure and function of opioid receptors, the structure-guided design of bitopic ligands is expected to lead a new wave of opioid drug development, advancing clinical pain management towards a more personalized and precise era.
In the GPCR receptors of the Class A family, the sodium ion binding site near D2.50 is considered as a classic allosteric regulatory site. This pocket undergoes significant conformational changes during receptor activation, which are crucial for the signal modulation of Class A GPCRs.
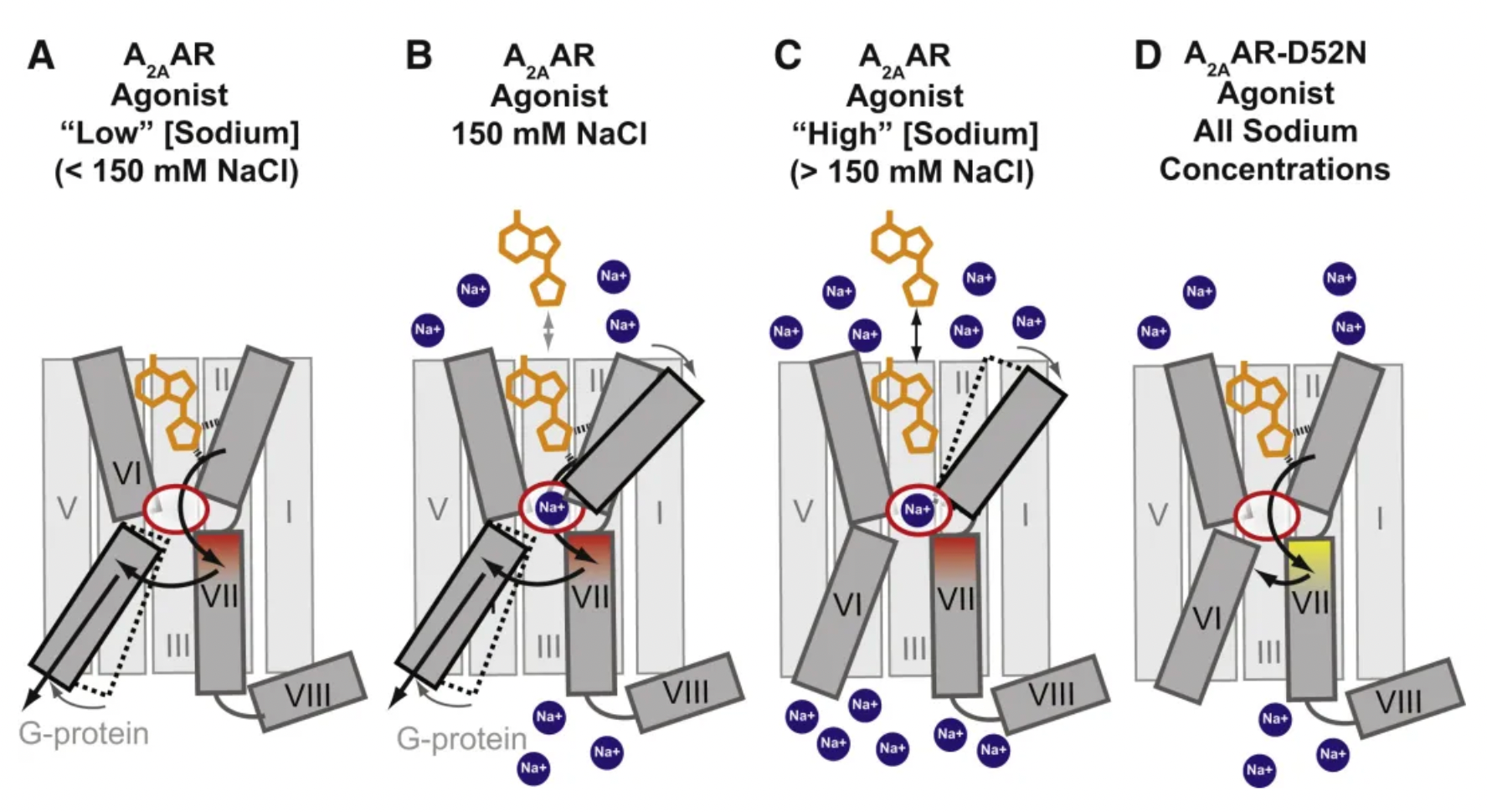
Previous studies have shown that the sodium ion binding site residues in opioid receptors act as a primary "efficacy switch," which can bias the downstream signaling towards either the Gi protein or the β-arrestin-2 pathway. The high-resolution structure of the μ-opioid receptor (μOR) in complex with the agonist Bu72 (PDB ID: 5C1M) reveals conformational changes associated with activation within this cavity, which retains several water molecules.
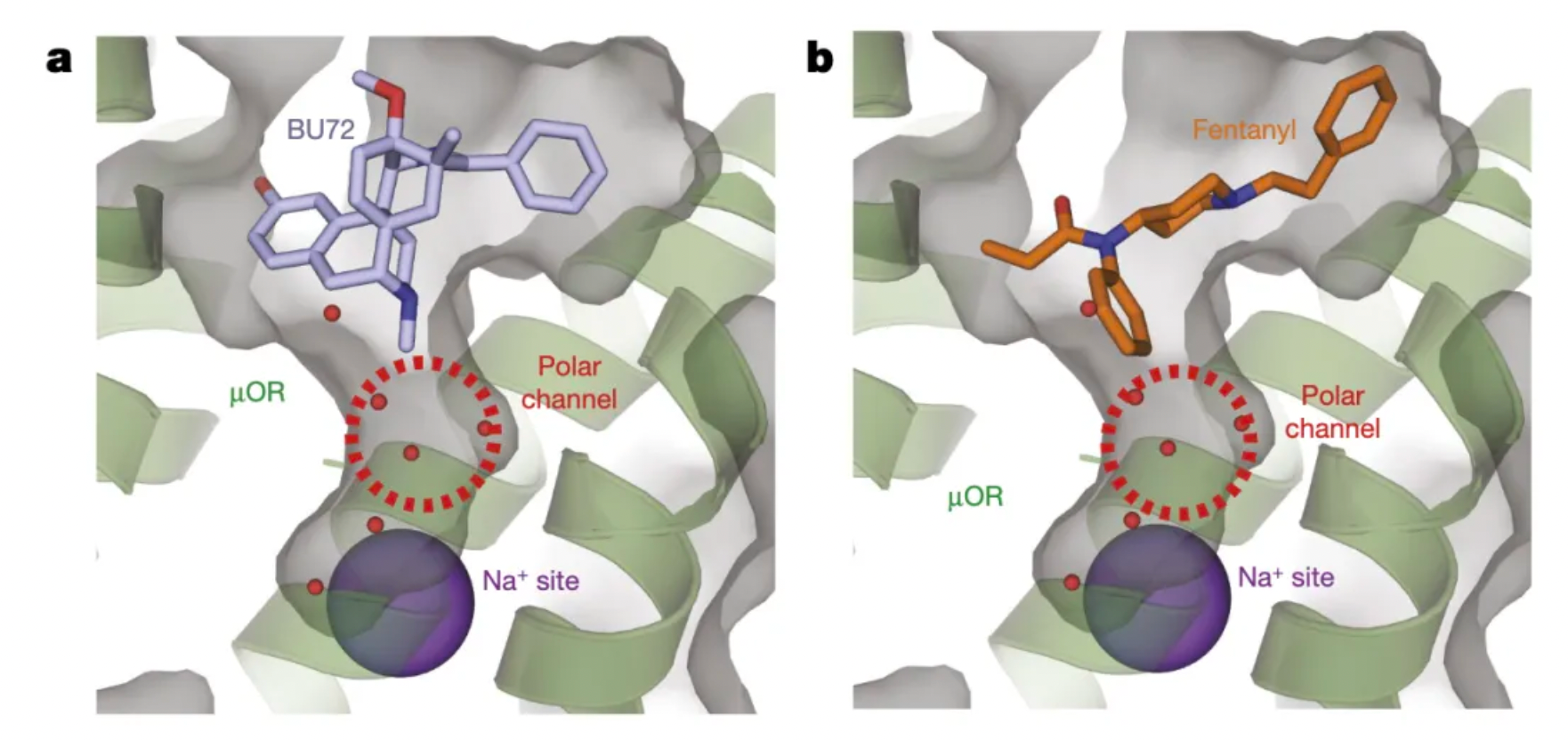
Researchers, starting with the structure of fentanyl, aimed to design drugs to explore the role of the cavity between the orthosteric binding site and the sodium ion binding site in receptor conformational regulation. By adjusting the length of different linkers and the type of terminal positively charged moieties, the researchers separately investigated the biasing differences in agonist activity at the μ-opioid receptor (μOR) with amines and guanidines in different linkers.
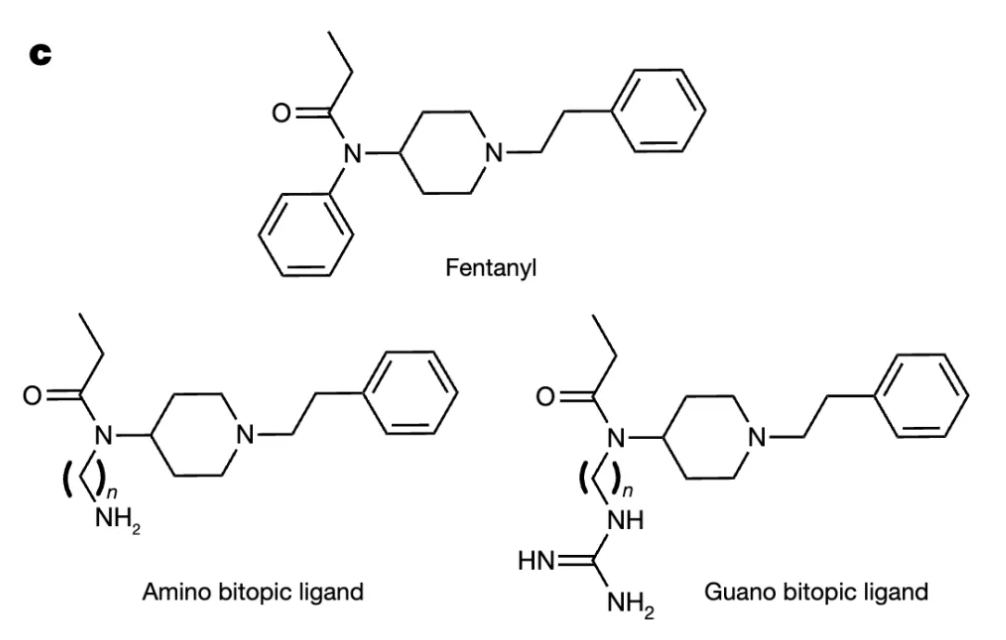
The results of molecular docking of the redesigned compounds indicated that the positively charged amines or guanidines were capable of extending into the classic sodium ion binding site and forming salt-bridge interactions with the key amino acid residue D2.50.
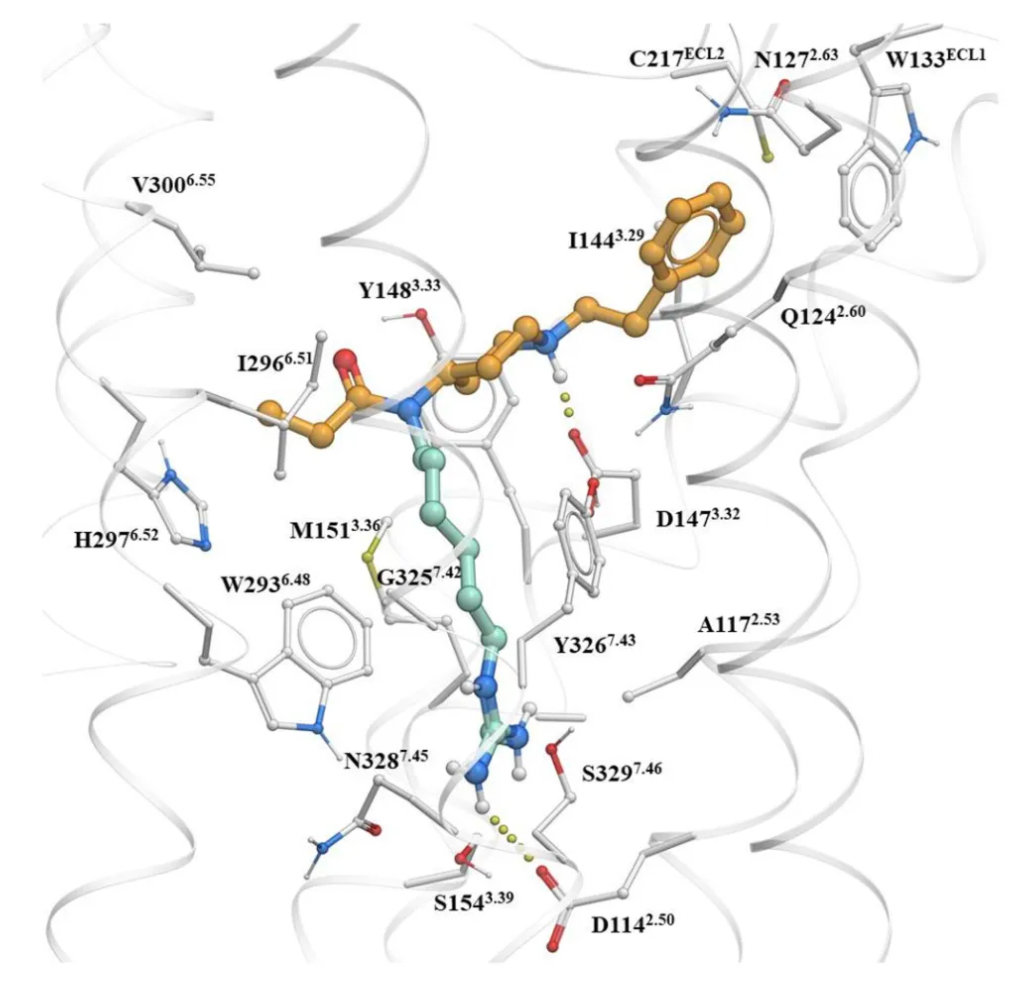
The table below presents the activity profiles of compounds formed by combining linkers of different lengths with amino and guanidino groups. It is evident that the affinity of amino-fentanyl derivatives for the μ-opioid receptor (μOR) is reduced, indicating that the amino head group does not effectively interact with the polar and negatively charged residues of the sodium ion-binding pocket (D114^2.50, N150^3.35, N328^7.45, S329^7.46, and S154^3.39). Furthermore, the extension of the alkyl chain does not appear to significantly affect the binding capability of such molecules, with the inhibitory constants (Ki values) for C9-amino and C7-amino ligands being 281 nM and 369 nM, respectively.
In contrast, all guanidino derivatives displayed moderate to high affinity for the μOR, with Ki values ranging from 590 nM for C11-guanidino derivatives to 4.6 nM and 4.1 nM for C5-guanidino and C6-guanidino, respectively. The latter showed a slight improvement in binding capability compared to fentanyl (Ki = 9 nM).
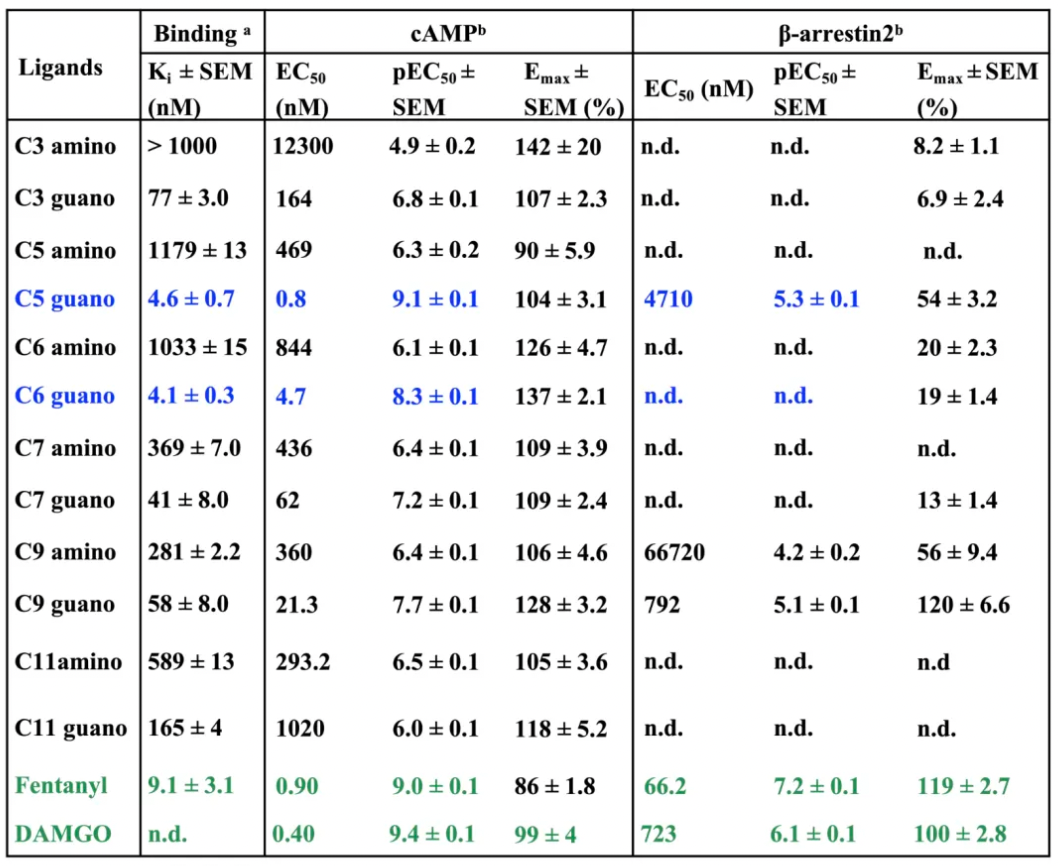
Bias functional selectivity analysis revealed that C6-guanidino exhibited weaker β-arrestin2 recruitment efficiency compared to C5-guanidino, while also displaying a higher maximal effect (Emax) associated with the second messenger cAMP signaling pathway.
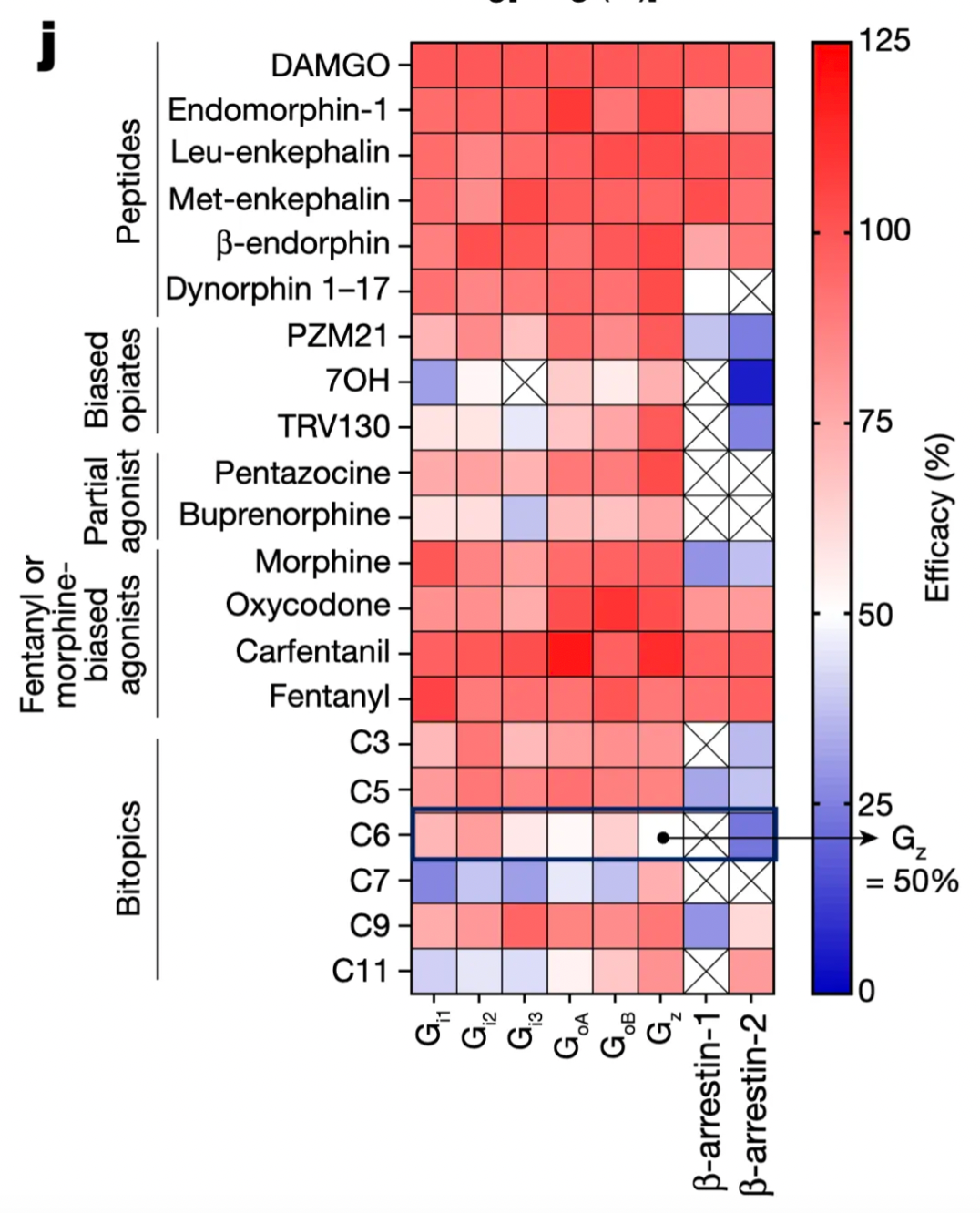
To further investigate the biased activation towards different G-protein subtypes and the two β-arrestin1/2 signaling pathways associated with cAMP inhibition, researchers conducted functional assays on six different G-proteins and β-arrestin1/2 signaling pathways. The results highlighted the particularly prominent functional selectivity of C6-guanidino, which maintained nanomolar potency and high activity in the Gi subtype, but demonstrated decreased efficacy in the Gz subtype. In the experiments, the researchers also tested the functional selectivity of various μOR agonists, including biased agonists, morphinan analogs, fentanyl analogs, full agonists (such as DAMGO, fentanyl, carfentanil, etc.), and G-protein-biased partial agonists.
In addition, researchers have investigated the effective analgesic action of C6-guanidine in animal models, and this molecule has shown a reduction in the typical adverse reactions mediated by μ-opioid receptors (μOR).




