Request Demo
Last update 19 Oct 2025
Insulin human (rDNA origin, Novo Nordisk)
Last update 19 Oct 2025
Overview
Basic Info
Drug Type Hormone |
Synonyms human insulin, human insulin (rDNA), insulin human + [33] |
Target |
Action agonists |
Mechanism INSR agonists(Insulin receptor agonists) |
Therapeutic Areas |
Active Indication |
Inactive Indication |
Originator Organization |
Active Organization |
Inactive Organization |
License Organization |
Drug Highest PhaseApproved |
First Approval Date United States (30 Aug 1983), |
Regulation- |



Login to view timeline
Structure/Sequence
Sequence Code 4857α

Sequence Code 4868β
Related
84
Clinical Trials associated with Insulin human (rDNA origin, Novo Nordisk)NCT06558422
Human Models of Selective Insulin Resistance: Pancreatic Clamp

NCT06434038
Measurement of Insulin Levels in the Cerebrospinal Fluid (CSF) of Healthy Adults After a Single Intranasal Dose - Middle Age
NCT07007130
Effect of Intraperitoneal Insulin Administration After Laparoscopy in Insulin-Resistant Patients on Prevention of Postoperative Adhesion Recurrence: A Randomized Controlled Trial

100 Clinical Results associated with Insulin human (rDNA origin, Novo Nordisk)
Login to view more data
100 Translational Medicine associated with Insulin human (rDNA origin, Novo Nordisk)
Login to view more data
100 Patents (Medical) associated with Insulin human (rDNA origin, Novo Nordisk)
Login to view more data
366
Literatures (Medical) associated with Insulin human (rDNA origin, Novo Nordisk)05 Sep 2025Journal of the Endocrine Society
Acute Effect of Intranasal Insulin on Food Intake Among Middle-Aged African American Adults: The FIINAAL Study
Article
Author: Manrique, Isabella R ; Goodson, Mia M ; Carmichael, Owen T ; Martin, Corby K ; Salceanu, Vanessa ; Rao, Arushi ; Beyl, Robbie ; Firmin, Simon ; Newton, Robert L ; Gwizdala, Kathryn L
Abstract:
Context:
Intranasal insulin has emerged as a promising potential treatment for cognitive decline. However, African American adults are under-represented in this research area, and unintentional weight loss is a possible detrimental side effect.
Objective:
We assessed effects of acute intranasal insulin exposure on food intake and appetite-related constructs among middle-aged, obese, cognitively normal African American adults. We hypothesized that intranasal insulin would result in fewer calories consumed, greater feelings of fullness, and less hunger compared to placebo.
Method:
A total of 39 participants received intranasal doses of Novolin-R (40 IU) and a saline placebo on separate days in a double-blind, counterbalanced, crossover design, with 3-day, eucaloric, nutritionally balanced diets preceding each dose. Doses were preceded by a 4-hour fast and followed by a test lunch. Visual analog scales (VAS) were used to assess appetite immediately before and after each dose, and after each lunch. Mixed effects linear model t tests were used to compare questionnaires and lunch intake between insulin and placebo.
Results:
There were no significant differences in food intake between conditions. However, feelings of fullness were significantly greater immediately after insulin compared to placebo. In addition, the desire to consume sweet foods decreased significantly more after insulin than after placebo.
Conclusion:
Acute intranasal insulin was associated with a reduced desire for sweet foods and with increased feelings of fullness, but not reduced food intake, among middle-aged African American adults. Eating behavior and appetite changes should be explored further as possible side effects of intranasal insulin treatment for cognitive decline in diverse populations.
01 Aug 2025JOURNAL OF CLINICAL PHARMACOLOGY
Comparing the Efficacy of Various Insulin Types: Pharmacokinetic and Pharmacodynamic Modeling of Glucose Clamp Effects in Healthy Volunteers
Article
Author: Chang, Yi Chien ; Jusko, William J.
Abstract:
This study compares the pharmacokinetics and efficacy of various subcutaneously (SC) dosed insulin analogs, including rapid‐acting, intermediate‐acting, long‐acting, and regular human insulin, using mechanistic pharmacokinetic (PK) and pharmacodynamic (PD) models. These models were applied to data from euglycemic clamp studies in healthy volunteers, where insulin pharmacokinetics and its effects on glucose utilization were monitored. Data from published studies were digitized and modeled using MONOLIX (Version 2024). The PK model described insulin absorption via sequential first‐order processes and linear elimination. The PD effects were captured using a model combination of biophase, indirect, and receptor down‐regulation components. While PK parameters—especially absorption rates—varied between insulin types, a common set of nonlinear PD parameters was sought to account for dose‐related differences in glucose utilization. The maximum glucose stimulation () was 163, and the insulin concentration for a half‐maximal effect () were 1156 pmol/L for insulin lispro, regular human insulin, neutral protamine hagedorn (NPH) insulin, and insulin glargine; 674 pmol/L for insulin aspart; and 5335 pmol/L for insulin detemir. Insulin detemir showed similar overt effects as the other insulin types but with smaller clearances and lower potency. This mechanism‐based glucose–insulin model demonstrated that most insulin analogs exhibit similar receptor‐ and transporter‐related parameters. The model, with specific PK but unified PD parameters, may enable clinical optimization of insulin therapy by highlighting differences in pharmacokinetics and operating common intrinsic glucose utilization parameters.
01 Aug 2025ANESTHESIA AND ANALGESIA
In Vitro Investigation of Insulin Dynamics During 4 Hours of Simulated Cardiopulmonary Bypass
Article
Author: Schweizer, Thilo ; Schild, Christof ; Bally, Lia ; Vogt, Andreas ; Guensch, Dominik P. ; Nagler, Michael ; Galova, Barbara ; Nossen, Caroline M. ; Fischer, Kady ; Huber, Markus ; Siepe, Matthias
BACKGROUND::
Hyperglycemia is common in patients undergoing cardiovascular surgery with cardiopulmonary bypass. We hypothesize that intraoperative hyperglycemia may be, at least partially, attributable to insulin loss due to adhesion on artificial surfaces and/or degradation by hemolysis. Thus, our primary aim was to investigate the loss of insulin in 2 different isolated extracorporeal circulation circuits (ECCs), that is, a conventional ECC (cECC) with a roller pump, and a mini-ECC (MiECC) system with a centrifugal pump. The secondary aim was to assess and compare the relationship between changes in insulin concentration and the degree of hemolysis in our 2 ECC models.
METHODS::
Six cECC and 6 MiECC systems were primed with red packed blood cells and thawed fresh-frozen plasma (1:1). Four additional experiments were performed in cECC using only thawed fresh-frozen plasma. Human insulin (Actrapid) was added, targeting a plasma insulin concentration of 400 mU/L. Insulin concentration and hemolysis index were measured at baseline and hourly thereafter. The end points were the change in insulin level after 4 hours compared to baseline and hemolysis index after 4 hours. The insulin concentration and hemolysis index were analyzed by means of a saturated linear mixed-effect regression model with a random offset for each experiment to account for the repeated measure design of the study, resulting in mean estimates and 95% confidence intervals (CIs) of the primary end points as well as of pairwise contrasts with respect to ECC type.
RESULTS::
Insulin concentration decreased by 63% (95% CI, 48%–77%) in the MiECC and 92% (95% CI, 77%–106%) in the cECC system that contained red blood cells. Insulin loss was significantly higher in the cECC system compared to the MiECC (P = .022). In the cECC with only plasma, insulin did not significantly decrease (−4%; 95% CI, −21% to 14%). Hemolysis index in MiECC increased from 68 (95% CI, 46–91) to 76 (95% CI, 54–98) after 4 hours, in cECC from 81 (95% CI, 59–103) to 121 (95% CI, 99–143). Hemolysis index and percent change of insulin showed an excellent relationship (r = −0.99, P < .01).
CONCLUSIONS::
Our data showed that insulin levels substantially decreased during 4 hours of simulated cardiopulmonary bypass only in the ECC that contained hemoglobin. The decrease was more pronounced in the cECC, which also exhibited a greater degree of hemolysis. Our results suggest that insulin degradation by hemolysis products may be a stronger contributor to insulin loss than adhesion of insulin molecules to circuit surfaces.
24
News (Medical) associated with Insulin human (rDNA origin, Novo Nordisk)
22 Apr 2025

AcquisitionDrug Approval
17 Feb 2025

Drug ApprovalBiosimilar
100 Deals associated with Insulin human (rDNA origin, Novo Nordisk)
Login to view more data
R&D Status
Approved
10 top approved records. to view more data
Login
| Indication | Country/Location | Organization | Date |
|---|---|---|---|
| Diabetes Mellitus | United States | 30 Aug 1983 |
Developing
10 top R&D records. to view more data
Login
| Indication | Highest Phase | Country/Location | Organization | Date |
|---|---|---|---|---|
| Shock, Septic | Phase 3 | Germany | 01 Apr 2003 | |
| Shock, Septic | Phase 3 | Germany | 01 Apr 2003 | |
| Shock, Septic | Phase 3 | Germany | 01 Apr 2003 | |
| Diabetes Mellitus, Type 1 | Phase 3 | United States | 01 Jun 2002 | |
| Diabetes Mellitus, Type 2 | Phase 2 | United States | 06 Oct 2015 | |
| Sepsis | Clinical | United States | 01 Jul 2003 | |
| Surgical Wound Infection | Clinical | United States | 01 Jul 2003 |
Login to view more data
Clinical Result
Clinical Result
Indication
Phase
Evaluation
View All Results
Phase 2 | 3 | iugfudtzwz = znmtjjsqir kjgsdhuxqy (qfwyxlwlcx, dmahanbpnp - fdalvutdsu) View more | - | 06 Jul 2023 | |||
Phase 2 | 289 | (Type 2 Diabetes Mellitus - Insulin) | oglvzgmqun(mrylozgwor) = cbtvkjbytr nxlmjqhyty (xlzgfcijxh, 22.94) View more | - | 17 Sep 2021 | ||
Placebo (Type 2 Diabetes Mellitus - Placebo) | oglvzgmqun(mrylozgwor) = zbefzqiwti nxlmjqhyty (xlzgfcijxh, 21.04) View more | ||||||
Phase 4 | 3 | (NPH Insulin) | kkmzkxuolq(jobwmiypfw) = ctokhgldsf xectcjjdbb (qhwmmanybj, 81.0) View more | - | 11 Sep 2020 | ||
glargine+Insulin Aspart (Basal/Bolus Insulin) | kkmzkxuolq(jobwmiypfw) = xvybqbpdim xectcjjdbb (qhwmmanybj, 61.8) View more | ||||||
Phase 2 | 15 | Intranasal Insulin (Insulin) | wicjobpwor(oahmzyiwrl) = optphpmdtt cfabdddayf (wtaghokufs, oypmbmjfii - uiefjkagwr) View more | - | 23 Nov 2018 | ||
Intranasal Insulin (Placebo) | wicjobpwor(oahmzyiwrl) = zdlyfvtuuc cfabdddayf (wtaghokufs, xcnkebrfku - pxoqnjqnst) View more | ||||||
Phase 4 | 109 | Analogue insulin (insulin detemir and insulin aspart) | pytqphxhof(mfawtrhvlf) = gobnpefiai xcbnpjsqlb (uhcpfslahp ) View more | - | 16 Jan 2016 | ||
Human insulin (NPH-insulin and regular HI) | pytqphxhof(mfawtrhvlf) = kguqiksovm xcbnpjsqlb (uhcpfslahp ) | ||||||
Not Applicable | 102 | (Intensive Insulin Treatment) | grslzlenfk = dvftauypwa onshumkyrh (tommhppgpb, rqqvewcapj - mtbrhhumwe) View more | - | 21 Dec 2015 | ||
(Standard Insulin Treatment) | grslzlenfk = yzjjfdhwia onshumkyrh (tommhppgpb, eabwabuelf - bwjewjyjig) View more | ||||||
Phase 4 | 261 | Exuber+Human Insulin (Inhaled Human Insulin (Exubera®)) | ujhfdcsapd(tofluqgpnu) = suqwbgvlei lpdolleawb (cykzijbfaa, 1.19) View more | - | 21 Oct 2009 | ||
(Insulin Glargine (Lantus®)) | ujhfdcsapd(tofluqgpnu) = kizijhkmyh lpdolleawb (cykzijbfaa, 1.14) View more |
Login to view more data
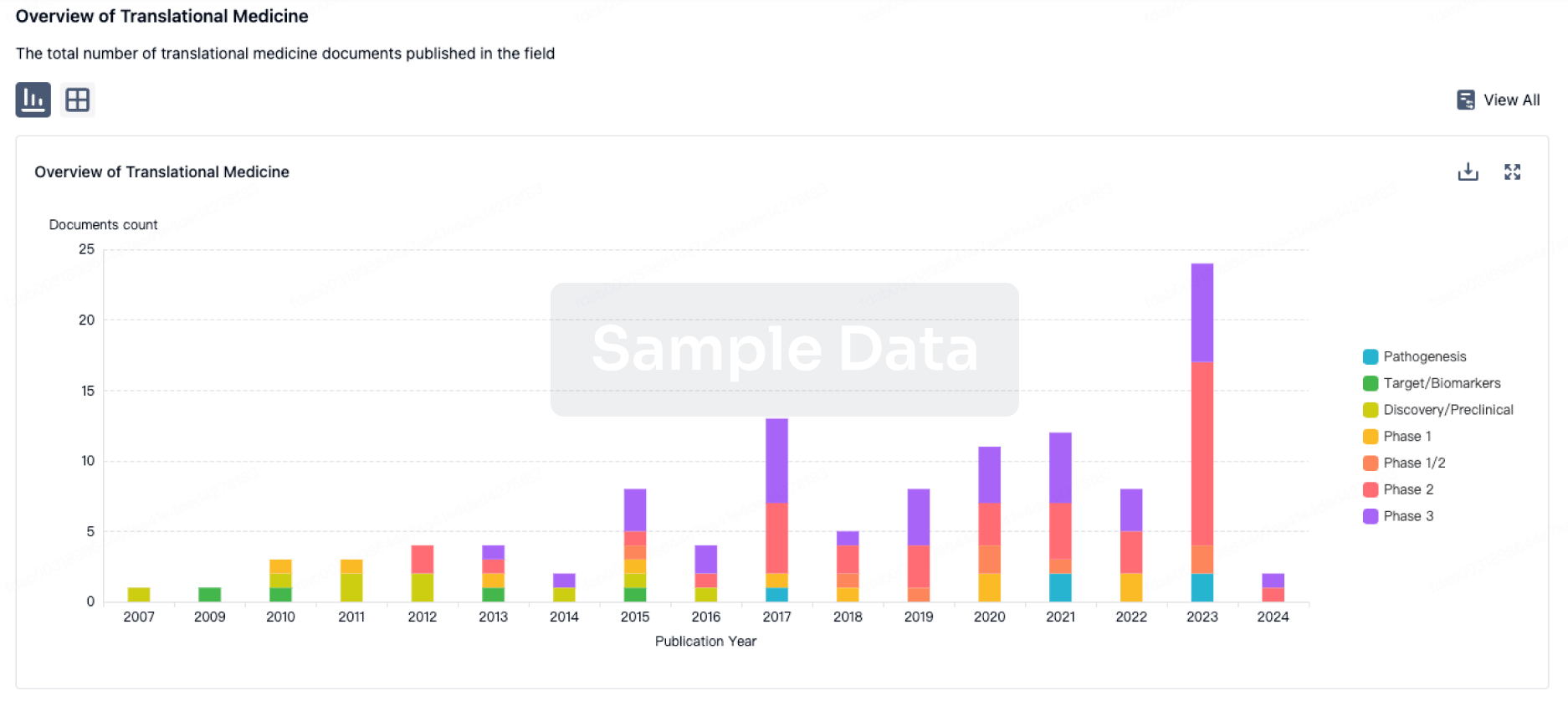
Translational Medicine
Boost your research with our translational medicine data.
login
or
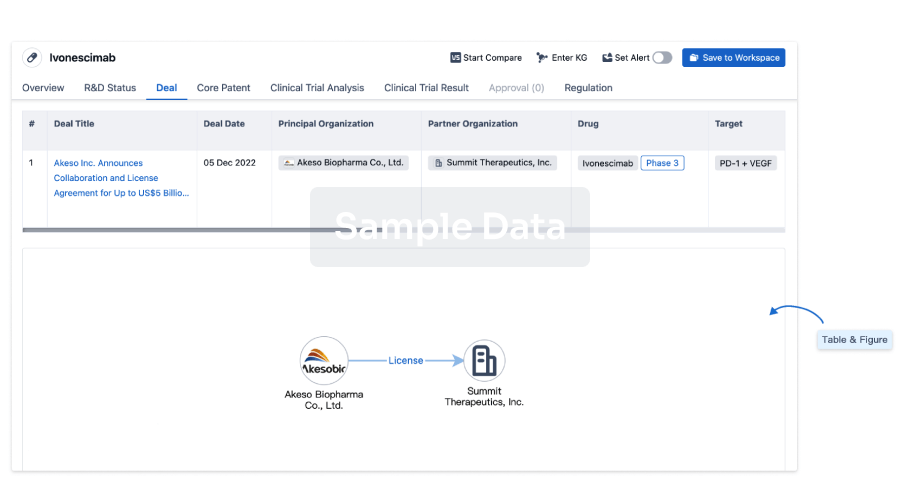
Deal
Boost your decision using our deal data.
login
or
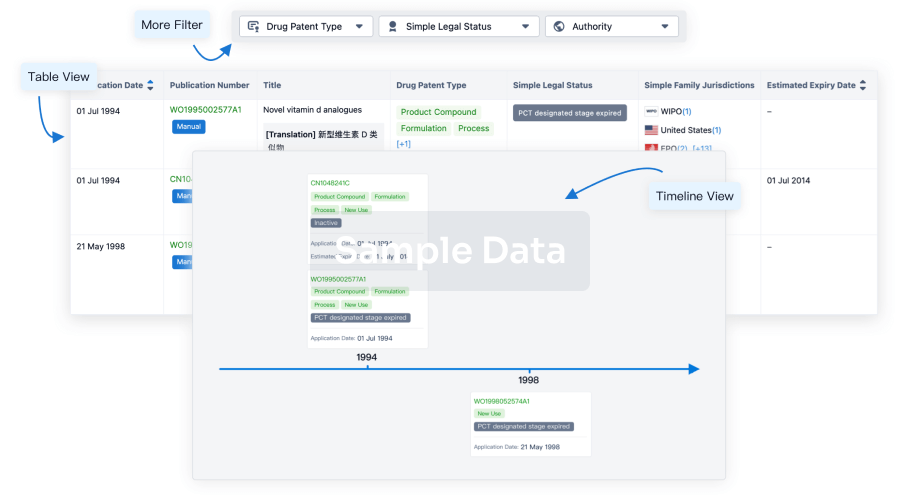
Core Patent
Boost your research with our Core Patent data.
login
or
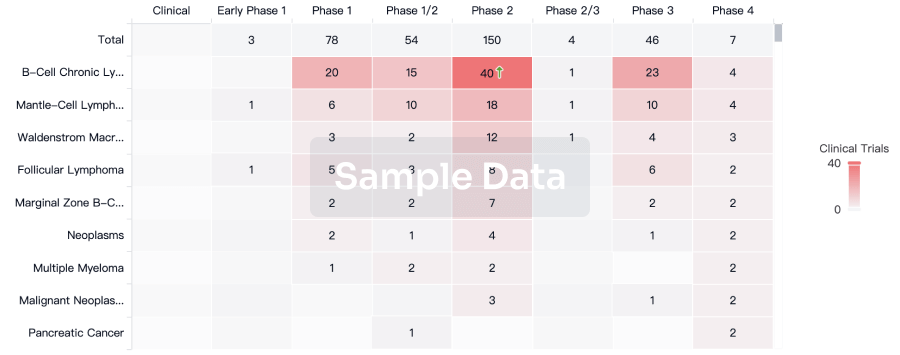
Clinical Trial
Identify the latest clinical trials across global registries.
login
or
Approval
Accelerate your research with the latest regulatory approval information.
login
or
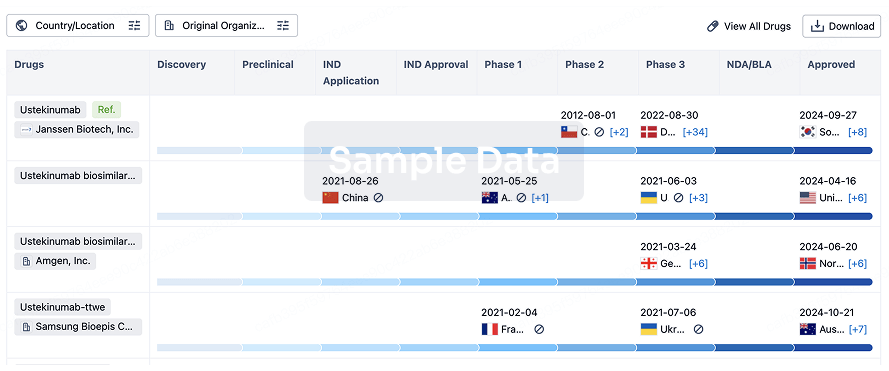
Biosimilar
Competitive landscape of biosimilars in different countries/locations. Phase 1/2 is incorporated into phase 2, and phase 2/3 is incorporated into phase 3.
login
or
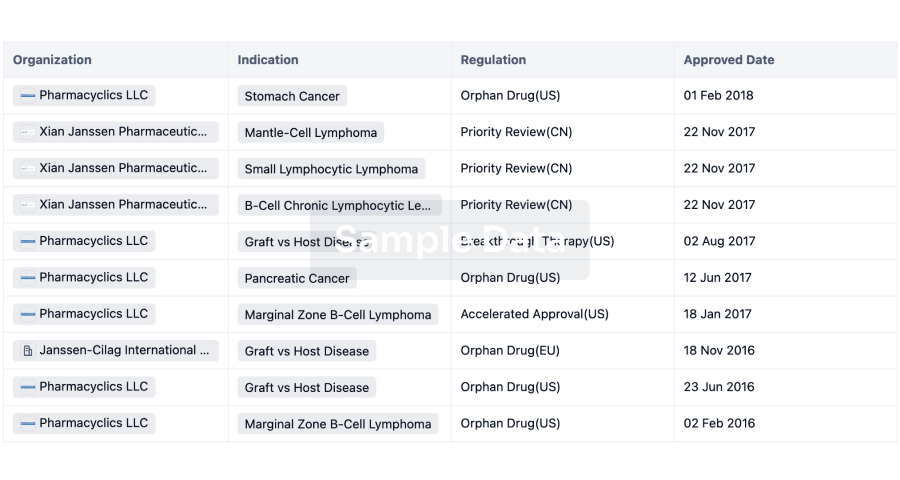
Regulation
Understand key drug designations in just a few clicks with Synapse.
login
or
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.
Bio
Bio Sequences Search & Analysis
Sign up for free
Chemical
Chemical Structures Search & Analysis
Sign up for free