Request Demo
Last update 09 Sep 2025
OPKO Health, Inc.
Last update 09 Sep 2025
Overview
Tags
Endocrinology and Metabolic Disease
Other Diseases
Nervous System Diseases
Hormone
CTP fusion protein
Synthetic peptide
Disease domain score
A glimpse into the focused therapeutic areas

Technology Platform
Most used technologies in drug development
Targets
Most frequently developed targets
Related
Target |
Mechanism |
Active Org. |
Originator Org. |
Active Indication |
Inactive Indication |
Drug Highest Phase |
First Approval Ctry. / Loc. |
First Approval Date |
Target |
Mechanism |
Active Org. |
Originator Org. |
Active Indication |
Inactive Indication |
Drug Highest Phase |
First Approval Ctry. / Loc. |
First Approval Date |

Target |
Mechanism |
Active Org. |
Originator Org. |
Active Indication |
Inactive Indication |
Drug Highest Phase |
First Approval Ctry. / Loc. |
First Approval Date |

NCT07110584
A Phase 1/2, Multi-Center, Open-Label Clinical Study Evaluating MDX2004 In Participants With Advanced Tumors

NCT06239194
A Phase 1/2a, Multicenter, First-in-human, Open-label Clinical Trial Evaluating MDX2001 Monotherapy in Patients With Advanced Solid Tumors
CTR20221404
一项评估IDX-1197联合XeIox(卡培他滨和奥沙利铂)或伊立替康用于晚期胃癌患者的安全性、耐受性和疗效的开放性、国际、多中心、Ib期研究
[Translation] An open-label, international, multicenter, phase Ib study evaluating the safety, tolerability, and efficacy of IDX-1197 in combination with XeIox (capecitabine and oxaliplatin) or irinotecan in patients with advanced gastric cancer

100 Clinical Results associated with OPKO Health, Inc.
Login to view more data
Login to view more data
09 Oct 2024Science Translational Medicine
A multispecific antibody against SARS-CoV-2 prevents immune escape in vitro and confers prophylactic protection in vivo
Article
Author: Kwong, Peter D. ; Zhang, Yi ; Oloniniyi, Olamide K. ; Boruszczak, Marika ; Andersen, Hanne ; Yang, Zhi-Yong ; Douek, Daniel C. ; Harris, Darcy R. ; Pegu, Amarendra ; Wei, Ronnie R. ; Wang, Lingshu ; Choe, Misook ; Porto, Maciel ; Godbole, Sucheta ; Shi, Wei ; Wei, Chih-Jen ; Zhao, Bingchun ; Moliva, Juan I. ; Wolff, Jeremy J. ; Suthar, Mehul S. ; Nabel, Gary J. ; Leung, Kwanyee ; Henry, Amy R. ; Zhou, Tongqing ; Olia, Adam S. ; Longobardi, Lindsay ; Carlton, Kevin ; Yang, Eun Sung ; Li, Juan ; Sullivan, Nancy J. ; Misasi, John ; Chen, Man ; Pessaint, Laurent ; Nase, Danielle ; Mascola, John R. ; Gall, Jason G. ; Lei, Q. Paula ; Ivleva, Vera B. ; Merriam, Jonah S. ; Koup, Richard A. ; Wallace, Shannon M. ; Liu, Cuiping
Despite effective countermeasures, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) persists worldwide because of its ability to diversify and evade human immunity. This evasion stems from amino acid substitutions, particularly in the receptor binding domain (RBD) of the spike protein that confers resistance to vaccine-induced antibodies and antibody therapeutics. To constrain viral escape through resistance mutations, we combined antibody variable regions that recognize different RBD sites into multispecific antibodies. Here, we describe multispecific antibodies, including a trivalent trispecific antibody that potently neutralized diverse SARS-CoV-2 variants and prevented virus escape more effectively than single antibodies or mixtures of the parental antibodies. Despite being generated before the appearance of Omicron, this trispecific antibody neutralized all major Omicron variants through BA.4/BA.5 at nanomolar concentrations. Negative stain electron microscopy suggested that synergistic neutralization was achieved by engaging different epitopes in specific orientations that facilitated binding across more than one spike protein. Moreover, a tetravalent trispecific antibody containing the same variable regions as the trivalent trispecific antibody also protected Syrian hamsters against Omicron variants BA.1, BA.2, and BA.5 challenge, each of which uses different amino acid substitutions to mediate escape from therapeutic antibodies. These results demonstrated that multispecific antibodies have the potential to provide broad SARS-CoV-2 coverage, decrease the likelihood of escape, simplify treatment, and provide a strategy for antibody therapies that could help eliminate pandemic spread for this and other pathogens.
01 Apr 2024Environmental research
Metal oxide nanomaterials based electrochemical and optical biosensors for biomedical applications: Recent advances and future prospectives
Review
Author: Kumar, Parveen ; Liu, Bo ; Huo, Peipei ; Behl, Gautam ; Upadhyaya, Kapil ; Rajan, Ramachandran ; Xiang, Xin-Xin
The amalgamation of nanostructures with modern electrochemical and optical techniques gave rise to interesting devices, so-called biosensors. A biosensor is an analytical tool that incorporates various biomolecules with an appropriate physicochemical transducer. Over the past few years, metal oxide nanomaterials (MONMs) have significantly stimulated biosensing research due to their desired functionalities, versatile chemical stability, and low cost along with their unique optical, catalytic, electrical, and adsorption properties that provide an attractive platform for linking the biomolecules, for example, antibodies, nucleic acids, enzymes, and receptor proteins as sensing elements with the transducer for the detection of signals or signal amplifications. The signals to be measured are in direct proportionate to the concentration of the bioanalyte. Because of their simplicity, cost-effectiveness, portability, quick analysis, higher sensitivity, and selectivity against a broad range of biosamples, MONMs-based electrochemical and optical biosensing platforms are exhaustively explored as powerful early-diagnosis tools for point of care applications. Herein, we made a bibliometric analysis of past twenty years (2004-2023) on the application of MONMs as electrochemical and optical biosensing units using Web of Science database and the results of which clearly reveal the increasing number of publications since 2004. Geographical area distribution analysis of these publications shows that China tops the list followed by the United States of America and India. In this review, we first describe the electrochemical and optical properties of MONMs that are crucial for the creation of extremely stable, specific, and sensitive sensors with desirable characteristics. Then, the biomedical applications of MONMs-based bare and hybrid electrochemical and optical biosensing frameworks are highlighted in the light of recent literature. Finally, current limitations and future challenges in the field of biosensing technology are addressed.
01 Apr 2024International journal of pharmaceutics
Design of liposomal nanocarriers with a potential for combined dexamethasone and bevacizumab delivery to the eye
Article
Author: Gasco, Paolo ; Fitzhenry, Laurence ; Behl, Gautam ; O'Reilly, Niall J ; Raghu Raj Singh, Thakur ; Ahmed, Zubair ; Farooq, Umer ; McLoughlin, Peter
Clinicians face numerous challenges when delivering medications to the eyes topically because of physiological barriers, that can inhibit the complete dose from getting to the intended location. Due to their small size, the ability to deliver drugs of different polarities simultaneously, and their biocompatibility, liposomes hold great promise for ocular drug delivery. This study aimed to develop and characterise a dual loaded liposome formulation encapsulating Bevacizumab (BEV) and Dexamethasone (DEX) that possessed the physicochemical attributes suitable for topical ocular delivery. Liposomes were prepared by using thin film hydration followed by extrusion, and the formulations were optimised using a design of experiments approach. Physicochemical characterisation along with cytocompatibility and bioactivity of the formulations were assessed. Liposomes were successfully prepared with a particle size of 139 ± 2 nm, PDI 0.03 ± 0.01 and zeta potential -2 ± 0.7 mV for the optimised formulation. BEV and DEX were successfully encapsulated into the liposomes with an encapsulation efficiency of 97 ± 0.5 % and 26 ± 0.5 %, respectively. A sustained release of BEV was observed from the liposomes and the bioactivity of the formulation was confirmed using a wound healing assay. In summary, a potential topical eye drop drug delivery system, which can co-load DEX and BEV was developed and characterised for its potential to be used in ocular drug delivery.
14 Aug 2025
Financial StatementExecutive Change
06 Aug 2025
VaccinemRNAExecutive Change
05 Aug 2025
VaccinemRNAExecutive Change
100 Deals associated with OPKO Health, Inc.
Login to view more data
100 Translational Medicine associated with OPKO Health, Inc.
Login to view more data
Corporation Tree
Boost your research with our corporation tree data.
login
or
Pipeline
Pipeline Snapshot as of 04 Dec 2025
The statistics for drugs in the Pipeline is the current organization and its subsidiaries are counted as organizations,Early Phase 1 is incorporated into Phase 1, Phase 1/2 is incorporated into phase 2, and phase 2/3 is incorporated into phase 3
Discovery
7
8
Preclinical
Phase 1
2
2
Phase 2
Phase 3
3
1
Approved
Other
41
Login to view more data
Current Projects
Login to view more data
Deal
Boost your decision using our deal data.
login
or
Translational Medicine
Boost your research with our translational medicine data.
login
or
Profit
Explore the financial positions of over 360K organizations with Synapse.
login
or
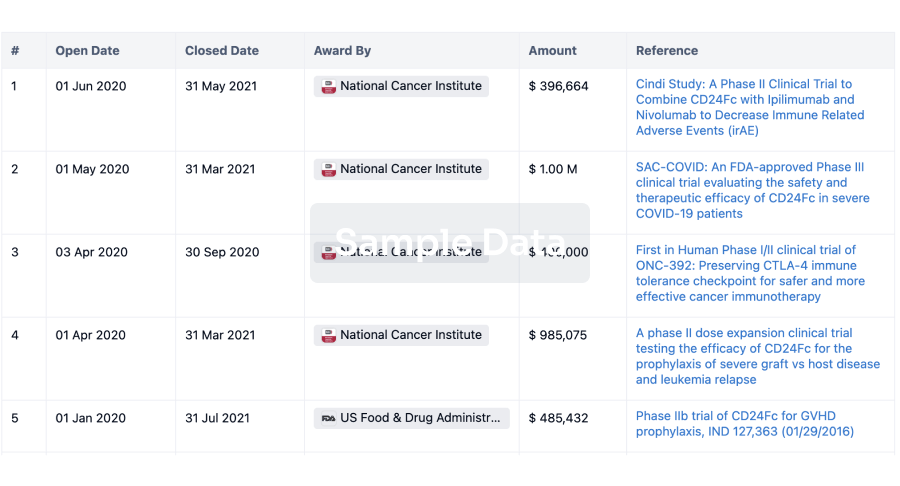
Grant & Funding(NIH)
Access more than 2 million grant and funding information to elevate your research journey.
login
or
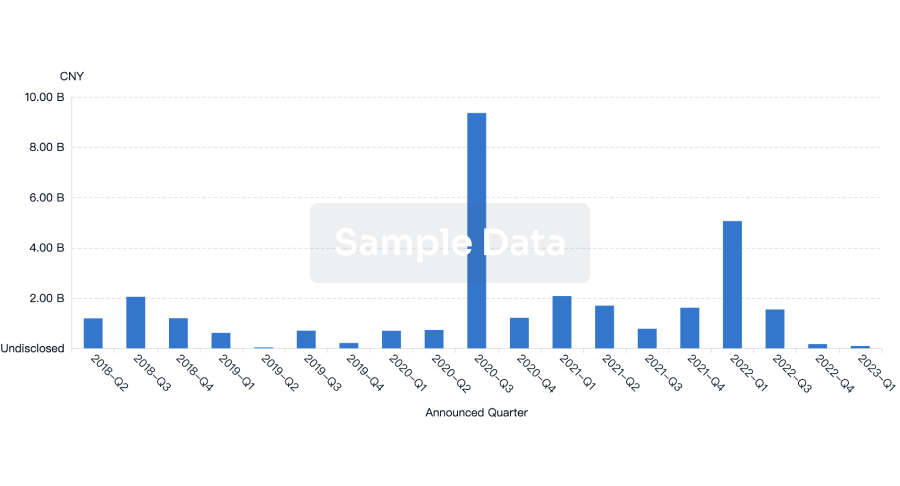
Investment
Gain insights on the latest company investments from start-ups to established corporations.
login
or
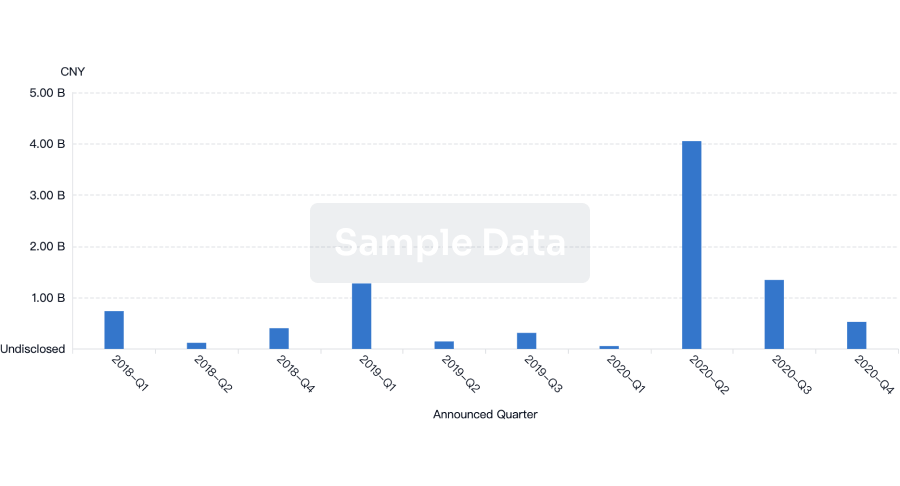
Financing
Unearth financing trends to validate and advance investment opportunities.
login
or
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.
Bio
Bio Sequences Search & Analysis
Sign up for free
Chemical
Chemical Structures Search & Analysis
Sign up for free