Request Demo
Last update 23 Jan 2025
PGE2 x calcium channel
Last update 23 Jan 2025
Basic Info
Related Targets |
Related
Target |
Mechanism |
Active Org. |
Originator Org. |
Active Indication |
Inactive Indication |
Drug Highest Phase |
First Approval Ctry. / Loc. |
First Approval Date |
100 Clinical Results associated with PGE2 x calcium channel
Login to view more data
100 Translational Medicine associated with PGE2 x calcium channel
Login to view more data
Login to view more data
01 Jan 2025Journal of Photochemistry and Photobiology B: Biology
The synergistic effect of pulsed red light and leonurus inhibits primary dysmenorrhea induced by oxytocin in mice by modulating calcium signaling and inhibiting inflammatory responses
Article
Author: Yang, Jiali ; Liu, Muqing ; Jiang, Hui ; Huo, Longfei ; Qin, Haokuan ; Lin, Shangfei ; Ren, Yi ; Fu, Qiqi ; Yao, Jinghui
01 May 2024Journal of Ethnopharmacology
Schisandrin B from Schisandra chinensis alleviated pain via glycine receptors, Nav1.7 channels and Cav2.2 channels
Article
Author: Wu, Jun ; Jin, Yuchen ; Li, Min ; Yu, KeXin ; Yu, Haibo ; Zhao, Miao
01 Jan 2023Biochemical and Biophysical Research Communications
Opioid modulation of T-type Ca2+ channel-dependent neuritogenesis/neurite outgrowth through the prostaglandin E2/EP4 receptor/protein kinase A pathway in mouse dorsal root ganglion neurons
Article
Author: Tsubota, Maho ; Yoshida, Shigeru ; Yamagata, Ryosuke ; Mitani, Kenji ; Sekiguchi, Fumiko ; Kawabata, Atsufumi ; Maeda, Takashi
Analysis
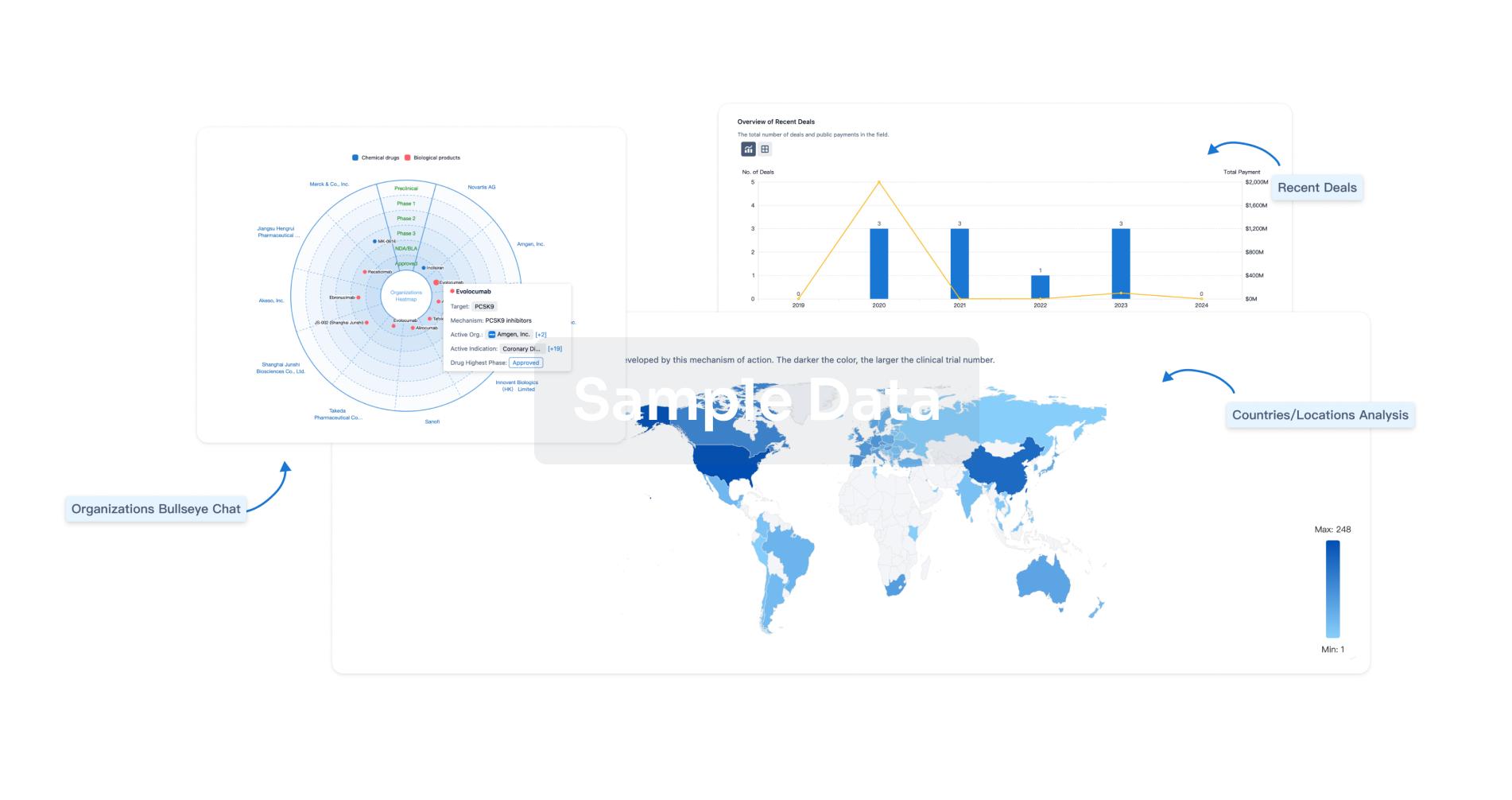
Perform a panoramic analysis of this field.
login
or
Chat with Hiro
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.
Bio
Bio Sequences Search & Analysis
Sign up for free
Chemical
Chemical Structures Search & Analysis
Sign up for free