Request Demo
What are Sickle haemoglobin modulators and how do they work?
21 June 2024
Sickle cell disease (SCD) is a genetic blood disorder characterized by the presence of abnormal hemoglobin, known as hemoglobin S (HbS). This abnormal hemoglobin causes red blood cells to assume a sickle or crescent shape, leading to various complications, including severe pain, anemia, and organ damage. While there is no universal cure for SCD, advancements in medical research have led to the development of sickle hemoglobin modulators, a promising class of therapeutic agents designed to manage the condition more effectively.
Sickle hemoglobin modulators work by targeting the underlying pathophysiology of SCD, primarily focusing on the abnormal hemoglobin and sickling of red blood cells. These modulators can act through various mechanisms, including increasing fetal hemoglobin (HbF) production, reducing hemoglobin S polymerization, and enhancing red blood cell hydration and deformability.
One of the primary approaches involves increasing the levels of fetal hemoglobin (HbF) in patients with SCD. HbF is a form of hemoglobin that is normally present in fetuses and newborns but decreases after birth. Unlike hemoglobin S, HbF does not form polymers and does not cause red blood cells to sickle. Some sickle hemoglobin modulators, such as hydroxyurea, work by stimulating the production of HbF. Higher levels of HbF can dilute the concentration of HbS within red blood cells, reducing the tendency for sickling and improving the overall function and lifespan of the cells.
Another approach focuses on reducing the polymerization of hemoglobin S. Polymerization is the process by which hemoglobin S molecules stick together to form long, rigid structures within red blood cells, leading to their sickling. Drugs like voxelotor work by binding to hemoglobin S and stabilizing it in a way that prevents polymerization. By inhibiting polymerization, these modulators can reduce the sickling of red blood cells and mitigate the associated complications.
Additionally, some sickle hemoglobin modulators aim to improve red blood cell hydration and deformability. In SCD, sickled red blood cells become rigid and less flexible, impairing their ability to navigate through small blood vessels. This can lead to blockages, reduced oxygen delivery to tissues, and subsequent pain and organ damage. Drugs such as crizanlizumab function by targeting cellular adhesion and inflammation, reducing the tendency of sickled cells to stick to blood vessel walls and each other. This can help maintain better blood flow and reduce the frequency of vaso-occlusive crises.
Sickle hemoglobin modulators are primarily used to alleviate the symptoms of SCD and prevent its complications. By stabilizing hemoglobin S, increasing fetal hemoglobin levels, and improving red blood cell properties, these drugs can significantly reduce the frequency and severity of vaso-occlusive crises—one of the hallmark complications of SCD. Vaso-occlusive crises are painful episodes that occur when sickled red blood cells obstruct blood flow, leading to tissue ischemia and severe pain.
Moreover, sickle hemoglobin modulators can help manage chronic anemia associated with SCD. Anemia in SCD results from the rapid destruction of sickled red blood cells, which have a much shorter lifespan than normal red blood cells. By reducing the sickling and destruction of red blood cells, these modulators can improve hemoglobin levels and alleviate the symptoms of anemia, such as fatigue and weakness.
Furthermore, the use of sickle hemoglobin modulators has shown promise in reducing the risk of long-term organ damage. Chronic organ damage is a significant concern in SCD, affecting the kidneys, liver, lungs, and other vital organs. By improving red blood cell function and reducing vaso-occlusive episodes, these drugs can help preserve organ function and improve the overall quality of life for patients with SCD.
In conclusion, sickle hemoglobin modulators represent a significant advancement in the management of sickle cell disease. By targeting the underlying mechanisms of hemoglobin S polymerization, increasing fetal hemoglobin levels, and improving red blood cell properties, these drugs offer a multifaceted approach to alleviating the symptoms and complications of SCD. While challenges remain, ongoing research and development in this field hold promise for even more effective treatments in the future, bringing hope to millions of individuals living with this debilitating condition.
Sickle hemoglobin modulators work by targeting the underlying pathophysiology of SCD, primarily focusing on the abnormal hemoglobin and sickling of red blood cells. These modulators can act through various mechanisms, including increasing fetal hemoglobin (HbF) production, reducing hemoglobin S polymerization, and enhancing red blood cell hydration and deformability.
One of the primary approaches involves increasing the levels of fetal hemoglobin (HbF) in patients with SCD. HbF is a form of hemoglobin that is normally present in fetuses and newborns but decreases after birth. Unlike hemoglobin S, HbF does not form polymers and does not cause red blood cells to sickle. Some sickle hemoglobin modulators, such as hydroxyurea, work by stimulating the production of HbF. Higher levels of HbF can dilute the concentration of HbS within red blood cells, reducing the tendency for sickling and improving the overall function and lifespan of the cells.
Another approach focuses on reducing the polymerization of hemoglobin S. Polymerization is the process by which hemoglobin S molecules stick together to form long, rigid structures within red blood cells, leading to their sickling. Drugs like voxelotor work by binding to hemoglobin S and stabilizing it in a way that prevents polymerization. By inhibiting polymerization, these modulators can reduce the sickling of red blood cells and mitigate the associated complications.
Additionally, some sickle hemoglobin modulators aim to improve red blood cell hydration and deformability. In SCD, sickled red blood cells become rigid and less flexible, impairing their ability to navigate through small blood vessels. This can lead to blockages, reduced oxygen delivery to tissues, and subsequent pain and organ damage. Drugs such as crizanlizumab function by targeting cellular adhesion and inflammation, reducing the tendency of sickled cells to stick to blood vessel walls and each other. This can help maintain better blood flow and reduce the frequency of vaso-occlusive crises.
Sickle hemoglobin modulators are primarily used to alleviate the symptoms of SCD and prevent its complications. By stabilizing hemoglobin S, increasing fetal hemoglobin levels, and improving red blood cell properties, these drugs can significantly reduce the frequency and severity of vaso-occlusive crises—one of the hallmark complications of SCD. Vaso-occlusive crises are painful episodes that occur when sickled red blood cells obstruct blood flow, leading to tissue ischemia and severe pain.
Moreover, sickle hemoglobin modulators can help manage chronic anemia associated with SCD. Anemia in SCD results from the rapid destruction of sickled red blood cells, which have a much shorter lifespan than normal red blood cells. By reducing the sickling and destruction of red blood cells, these modulators can improve hemoglobin levels and alleviate the symptoms of anemia, such as fatigue and weakness.
Furthermore, the use of sickle hemoglobin modulators has shown promise in reducing the risk of long-term organ damage. Chronic organ damage is a significant concern in SCD, affecting the kidneys, liver, lungs, and other vital organs. By improving red blood cell function and reducing vaso-occlusive episodes, these drugs can help preserve organ function and improve the overall quality of life for patients with SCD.
In conclusion, sickle hemoglobin modulators represent a significant advancement in the management of sickle cell disease. By targeting the underlying mechanisms of hemoglobin S polymerization, increasing fetal hemoglobin levels, and improving red blood cell properties, these drugs offer a multifaceted approach to alleviating the symptoms and complications of SCD. While challenges remain, ongoing research and development in this field hold promise for even more effective treatments in the future, bringing hope to millions of individuals living with this debilitating condition.
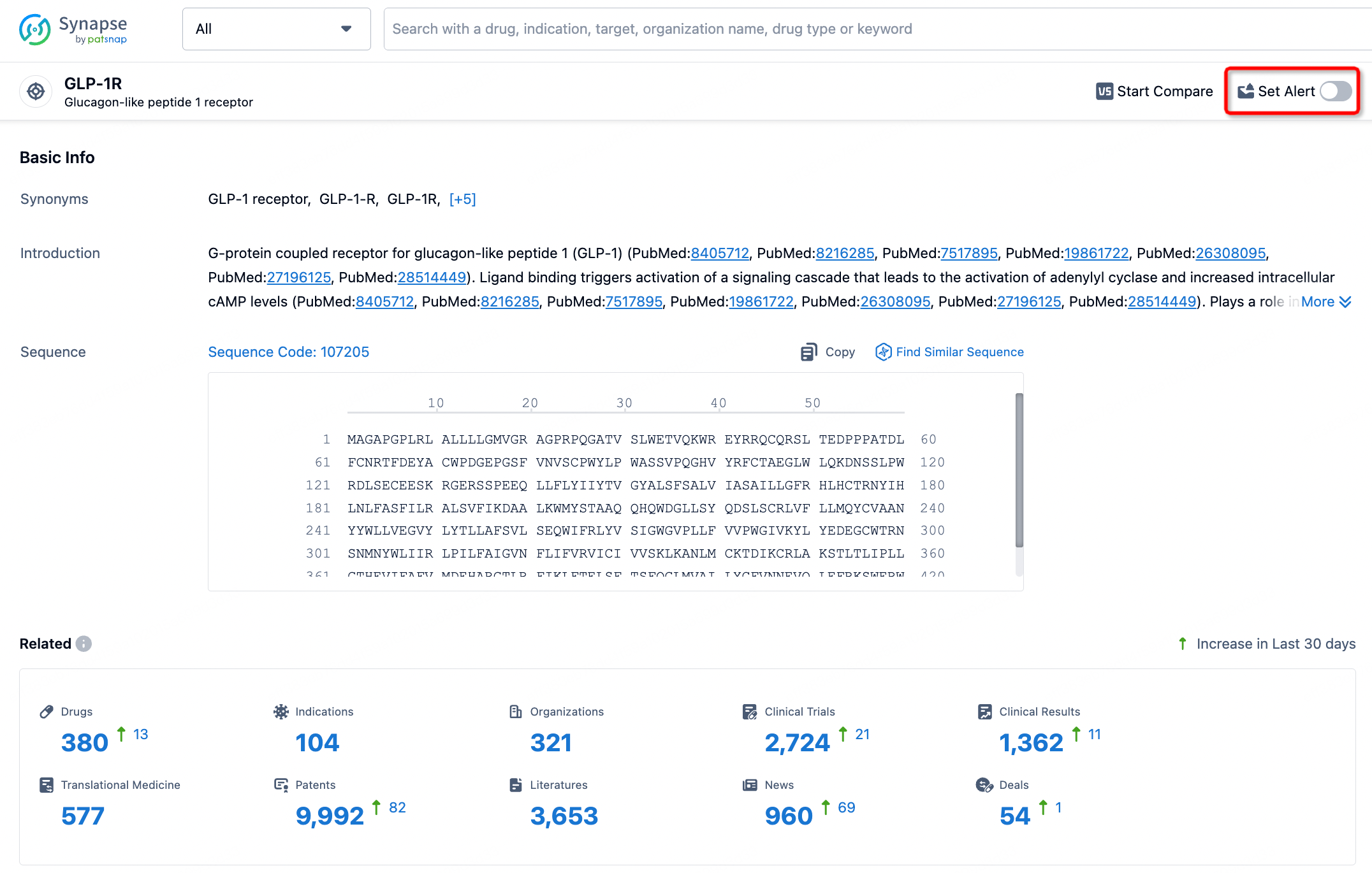
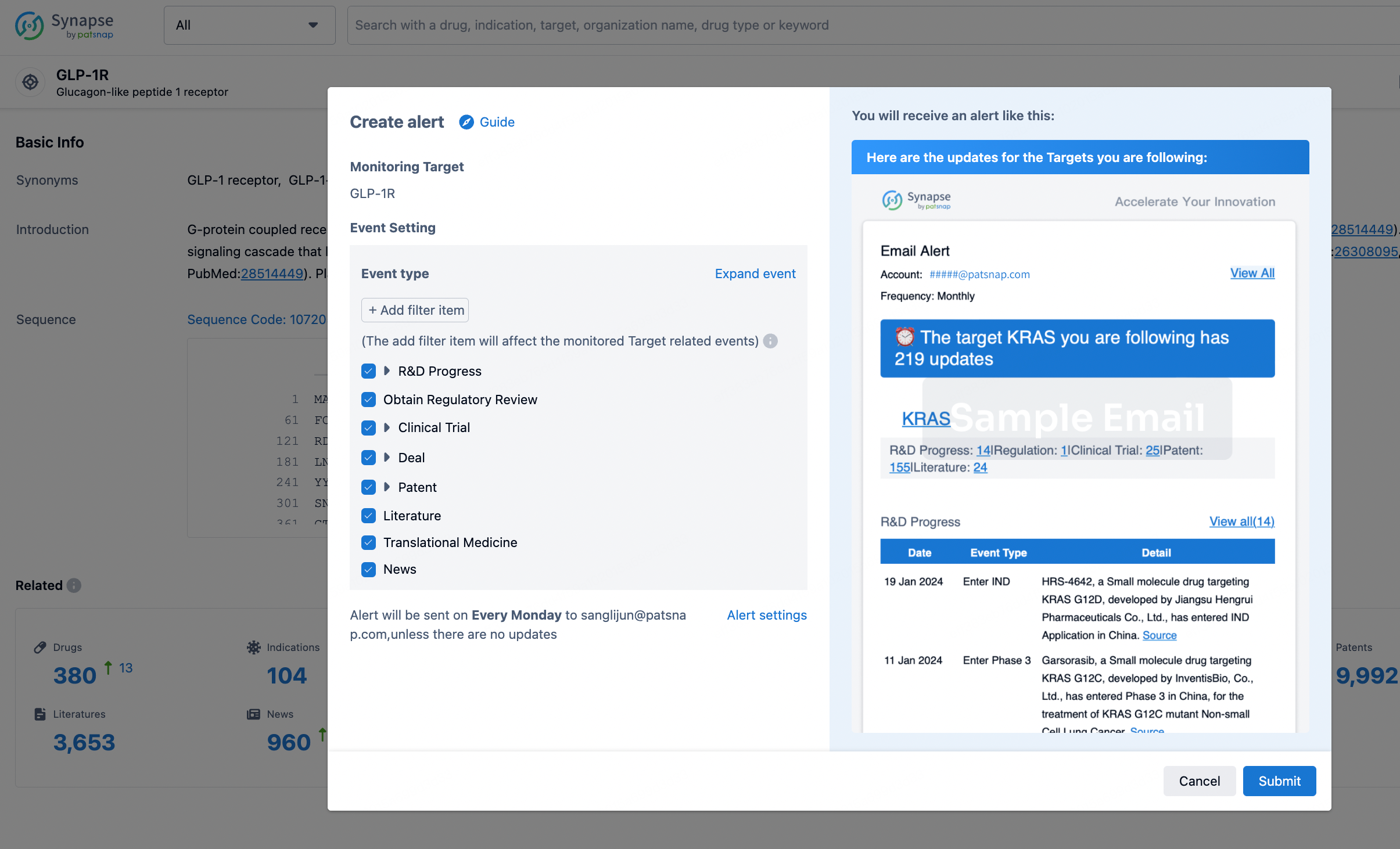
How to obtain the latest development progress of all targets?
In the Synapse database, you can stay updated on the latest research and development advances of all targets. This service is accessible anytime and anywhere, with updates available daily or weekly. Use the "Set Alert" function to stay informed. Click on the image below to embark on a brand new journey of drug discovery!
AI Agents Built for Biopharma Breakthroughs
Accelerate discovery. Empower decisions. Transform outcomes.
Get started for free today!
Accelerate Strategic R&D decision making with Synapse, PatSnap’s AI-powered Connected Innovation Intelligence Platform Built for Life Sciences Professionals.
Start your data trial now!
Synapse data is also accessible to external entities via APIs or data packages. Empower better decisions with the latest in pharmaceutical intelligence.