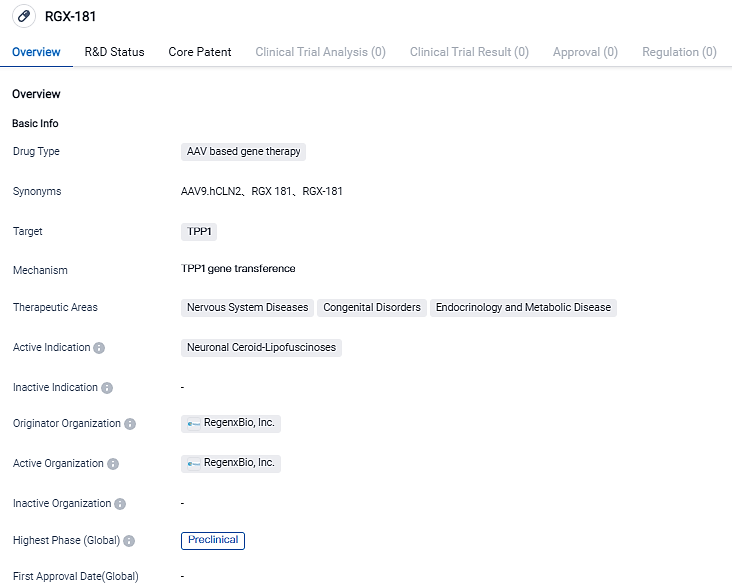
First Dosing of RGX-181 in a Pediatric CLN2 Patient's Initial Clinical Results Showcased at SSIEM Yearly Symposium
REGENXBIO Inc. shared the preliminary interim findings from a single-subject, researcher-led first-in-person study of RGX-181, designed to treat late-infantile neuronal ceroid lipofuscinosis type 2 (CLN2) disease, a variant of Batten disease, during the annual convention of Society for the Study of Inborn Errors of Metabolism (SSIEM) in Jerusalem.
👇Please click on the image below to directly access the latest data (R&D Status | Core Patent | Clinical Trial | Approval status in Global countries) of this drug.
"CLN2 is a debilitating condition caused by mutations in the gene known as CLN2, which leads to a deficiency of the TPP1 enzyme necessary for the digestion of certain peptides related to cellular waste. The symptoms of the disease, among other things, include seizures, deterioration of motor skills, language and cognitive capabilities, loss of vision, and early mortality. Current therapies have little effect in halting or reversing the majority of the disease's effects," according to REGENXBIO's Chief Health Officer, Steve Pakola, MD. "The initial findings imply that RGX-181 is safe and substantially reducing the patient's seizures who participated in this study, positing hope for progress."
Carolina Fischinger de Souza, Ph.D., M.D., the Hospital de Clínicas de Porto Alegre in Brazil, and the trial investigator remarked, "In my experience treating individuals suffering from this horrific disease, I've seen the deficiencies of existing treatments. The considerable reduction in seizure frequency, positive safety results, and decrease in ERT frequency are indicative of this gene treatment's potential to offer a significant treatment choice to the CLN2 community."
The first outcomes from a 5-year-old child who received a one-time intracisternal dosage of RGX-181 were reported today by a clinical investigation physician from the Hospital de Clinicas in Porto Alegre, Brazil. The follow-up period following administration lasted six months.
As of June 30, 2023, RGX-181 has been well received with no serious side effects. The treatment demonstrated steady TPP1 levels, greater gaps between Enzyme Replacement Therapy (ERT) infusions, and a decrease in seizure frequency by 86% over the six months, culminating in the discontinuation of two anti-epileptic drugs. The child also displayed promising advances in fine motor and expressive language skills.
Kenneth T. Mills, CEO and President of REGENXBIO, stated, "This third consecutive series of clinical trials of our treatment pipeline for rare neurodegenerative disease in our active clinical-stage demonstrates once again that potential one-time gene treatment is safe, re-establishes normal levels of gene expression and initiates significant changes in young patients. We have been granted crucial governing approvals for each of these three pipeline programs. We plan to submit our first Biologics License Application in 2024 for a rare neurodegenerative condition, Hunter syndrome, under the Fast-Track Acceptance procedure."
About RGX-181
RGX-181 aims to offer a novel, one-time remedy for CLN2 disease by utilizing the NAV AAV9 vector to distribute the gene that dictates TPP1, the missing enzyme in children with CLN2. The RGX-181 treatment is designed to provide patients with a continuous source of TPP1 post single intracisternal injection, potentially resulting in a long-lasting remediation of various cells in the CNS. In CLN2 animal trials, RGX-181 effectively restored TPP1 action to levels beyond those found in unaffected animals and improved neurological functionality and survival. RGX-181 could potentially serve as a valuable one-time therapeutic alternative for individuals with CLN2 based on the extensive CNS correction observed.
👇Please click on the picture link below for free registration or login directly if you have freemium accounts, you can browse the latest research progress on drugs , indications, organizations, clinical trials, clinical results, and drug patents related to this target.
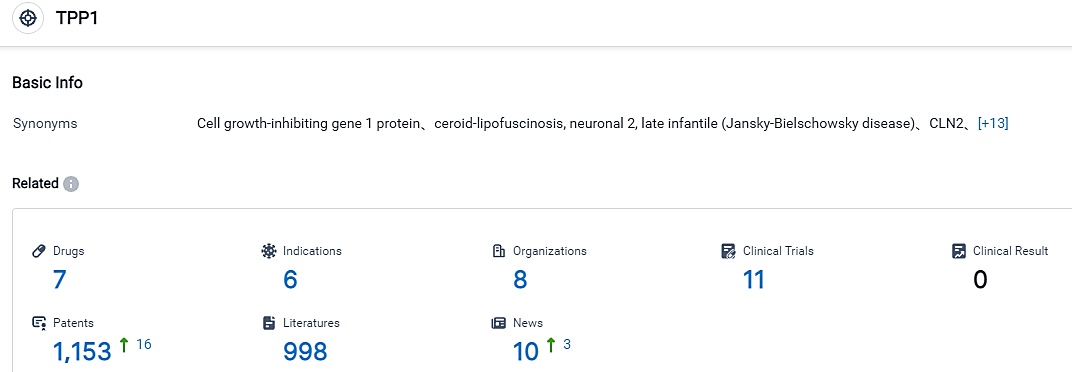
According to the information disclosed by the Synapse Database, as of September 1, 2023, there are 7 investigational drugs for the TPP1 target, including 6 applicable indications, 8 R&D institutions involved, with related clinical trials reaching 11, and as many as 1153 patents. Late-infantile neuronal ceroid lipofuscinosis type 2 disorder, also known as a variant of Batten disease, is a rare, juvenile-onset, autosomal recessive, neurodegenerative lysosomal storage condition induced by abnormalities in the TPP1 gene. A lack in TPP1 enzyme activity leads to a buildup of storage material in lysosomes and the deterioration of nerve cells, mostly in the brain and retina. CLN2 disorder features seizures, swift regression of language and motor capabilities, cognitive impairment, rapid vision loss leading to blindness, and early death by mid-childhood. Symptoms typically appear between the ages of two and four, initially presenting as repeated seizures (epilepsy), language retardation and difficulty in coordinating movements. At present, there is no known cure for CLN2 disorder. Available treatment methods consist of CNS enzyme replacement therapy, in which TPP1 is administered into the lateral ventricles through a non-removable device every two weeks, and palliative care. Currently, no approved therapies exist to address the eye-related symptoms of CLN2 disorder.




