Revolutionizing Drug Discovery: The Impact of Deep Learning Technologies in Life Sciences and Biotechnology
In today's field of life sciences and biotechnology, deep learning technology is reshaping the drug discovery process in unprecedented ways. With the rapid growth of computational power and the advent of the big data era, scientists have begun exploring how to leverage these new technologies to accelerate the drug development process. From AlphaFold, which predicts the three-dimensional structure of proteins, to ExPecto, which handles complex biomolecular structures and predicts drug-protein interactions, to DeepVariant, which identifies genomic variations using deep learning models, these tools are gradually changing our understanding and practice of life sciences.
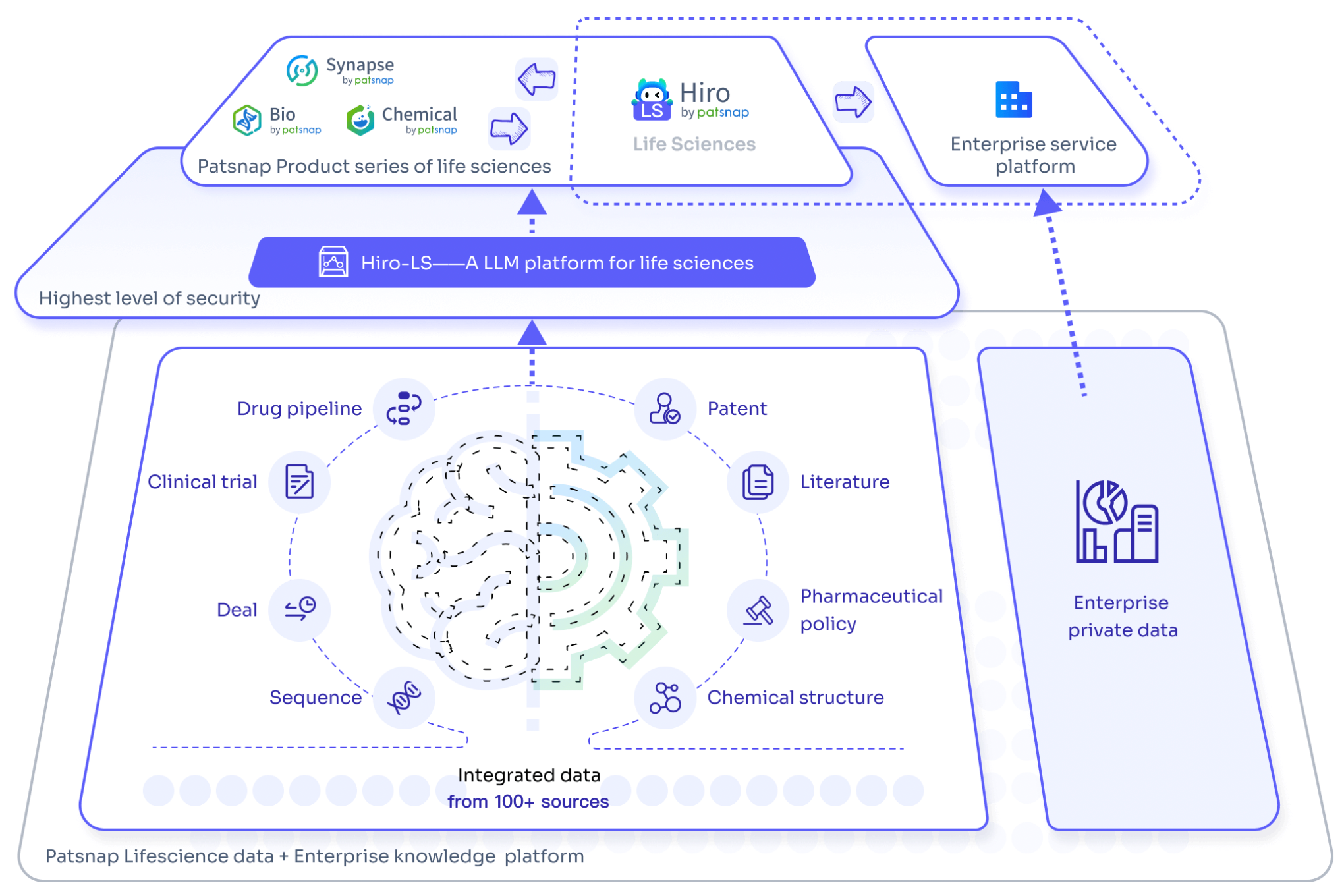
In an era characterized by information overload, effectively managing and utilizing vast amounts of biomedical data has become another pressing issue. Hiro-LS, launched by Patsnap, is a prime example of a solution to this challenge. Based on PharmaGPT, it can not only process complex biomedical data but also help users quickly access and understand key information via intelligent search and intuitive chart generation, thus driving innovation and development in the field of biomedicine.
Drug-Target Discovery Large Model—DeepPurpose
DeepPurpose is an open-source deep learning framework for drug discovery, developed by the research team at the University of Texas at Austin. This framework is designed to support predictions of drug-target interactions (DTI) and drug-disease associations (DDA). DeepPurpose utilizes deep learning technology to simulate interactions between drugs and targets, providing a powerful tool for drug discovery.
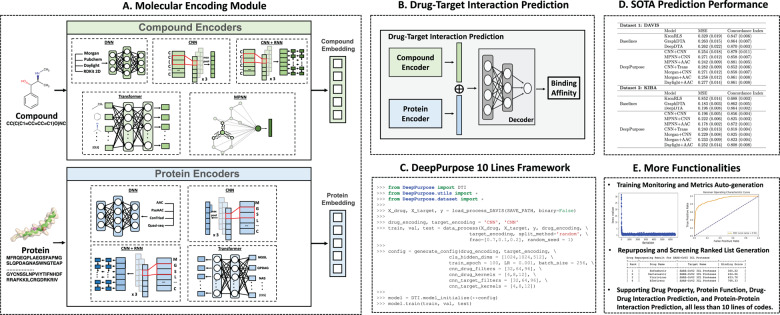
DeepPurpose offers a variety of compound and protein encoders, including traditional cheminformatics fingerprint encoders such as Morgan, PubChem, Daylight, and RDKit 2D fingerprints, as well as more modern deep learning models like Convolutional Neural Networks (CNN), Recurrent Neural Networks (RNN), Transformers, and Graph Neural Networks (GNN). The framework supports over 50 neural network architectures, allowing for the combination of different encoders to create highly customized models to meet specific research needs. DeepPurpose is designed to simplify the complex deep learning process, enabling researchers without extensive deep learning backgrounds to quickly get started. Its programming framework is clear and concise, requiring only a few lines of code to complete model training and predictions. In addition to Drug-Target Interaction (DTI) prediction, DeepPurpose also supports other tasks such as Drug-Drug Interaction (DDI), Protein-Protein Interaction (PPI), and protein function prediction. DeepPurpose provides a large collection of pre-trained models, which have been validated on multiple benchmark datasets and can be fine-tuned as base models to fit specific research tasks. Users can adjust model parameters, perform hyperparameter tuning, and employ methods like Bayesian optimization to find the optimal model configuration.
Drug-Protein Interaction Large Model - ExPecto
ExPecto is a model for predicting drug-protein interactions, created by the research team at the Korea Advanced Institute of Science and Technology (KAIST). Based on the Transformer architecture, it is capable of handling complex biomolecular structures and predicting interactions between drugs and proteins. This helps in identifying potential effective compounds during the drug design phase.
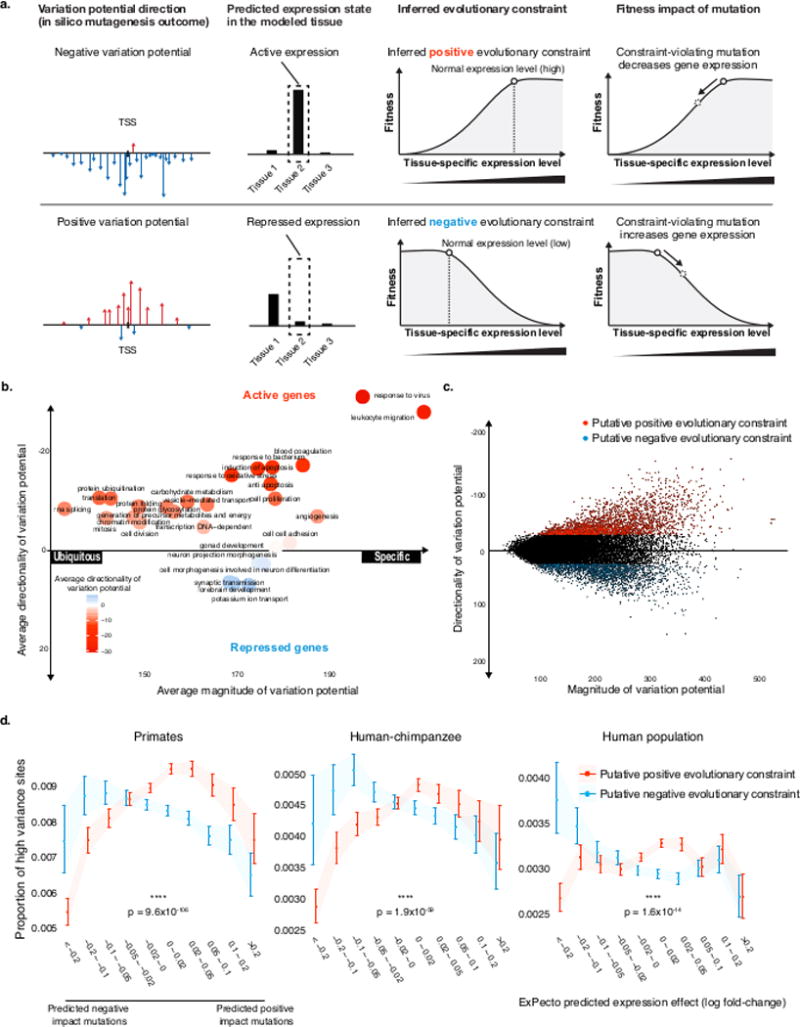
ExPecto combines advanced technologies such as Convolutional Neural Networks (CNN) and Gradient Boosting Decision Trees (GBDT) to assess how single nucleotide variants (SNVs) and insertion-deletion mutations (InDels) alter gene expression patterns, thereby revealing the potential biological and disease risks associated with these mutations. ExPecto utilizes deep learning techniques to handle complex genomic data, supporting the segmentation of large amounts of genetic data and enhancing its capability to process large-scale datasets. From variant detection to effect prediction, ExPecto achieves a highly automated workflow and allows users to train models using custom expression profiles according to specific needs. This tool plays a significant role in fields such as genetic disease research, personalized medicine, and bioinformatics education, helping researchers identify the associations between gene variants and diseases, thereby accelerating the development of new therapies or preventative measures.
Genetic Variant Detection Tool—DeepVariant
DeepVariant is a deep learning-based genetic variant detection tool developed by the Google Brain Genomics team. It was initially designed to identify single nucleotide polymorphisms (SNPs) and small insertions/deletions (indels) from next-generation sequencing (NGS) data. It works by converting aligned read data (typically stored in BAM or CRAM file formats) into a series of image tensors, which are then classified using a convolutional neural network (CNN), ultimately outputting standard VCF or gVCF files. The workflow of DeepVariant generally includes several steps: sample generation, sample encoding, network training, and generating variant calling results.
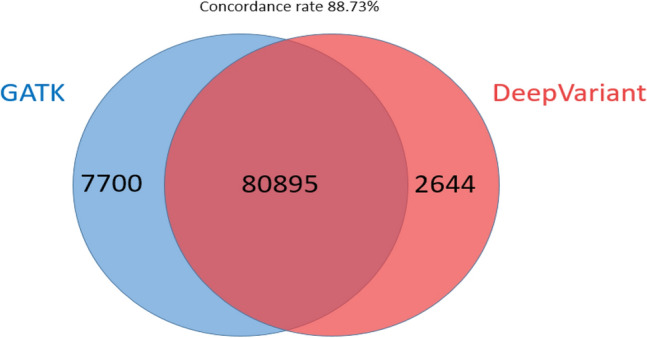
The core technology of DeepVariant lies in its deep learning model, which can accurately identify SNPs and small insertions/deletions from sequencing data. The workflow includes four main steps: data preprocessing, image generation, model classification, and output of results. DeepVariant's model, trained extensively on human data, exhibits high accuracy and robustness, performing exceptionally well in several international competitions. Originally designed for genomic variant detection, DeepVariant has been widely applied in biomedical research, particularly in gene editing technologies and genetic disease diagnostics. By enhancing the accuracy of genomic variant detection, DeepVariant has had a profound impact on personalized medicine and genetic research. Its flexibility and ease of use make it a powerful tool in scientific research and clinical applications. DeepVariant supports multiple sequencing technologies and data types, including Illumina, PacBio HiFi, and Oxford Nanopore. Furthermore, DeepVariant also supports the use of pangenome mappings to increase accuracy, making it a highly versatile tool in the field of bioinformatics.
Large Model for Protein 3D Structure Prediction—AlphaFold
Developed by DeepMind, AlphaFold is a deep learning model that predicts the three-dimensional structure of proteins. AlphaFold reconstructs the spatial conformation of proteins by predicting the distance distribution between amino acid residues. The core of AlphaFold is its end-to-end deep neural network, which extracts key features from amino acid sequences and further enriches the input through multiple sequence alignment (MSA) and template information. The success of AlphaFold means that scientists are now able to predict protein structures more accurately, which is crucial for drug design and understanding protein functions.
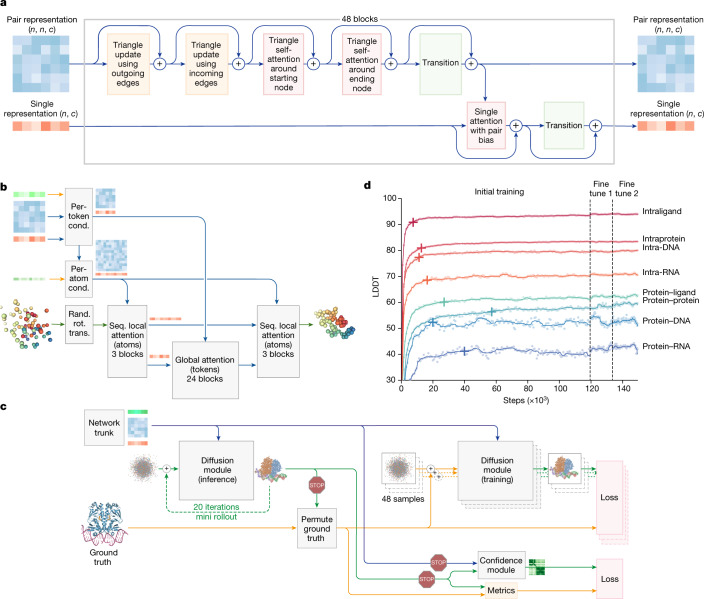
The working principle of AlphaFold is based on the conservation of protein structures through evolution. It utilizes an end-to-end deep learning network, which is trained to predict protein structures directly from amino acid sequences using information from homologous proteins and multiple sequence alignments. In AlphaFold, an attention-based Transformer architecture is employed to enhance its performance. Since demonstrating its outstanding predictive capability at CASP 14 (Critical Assessment of protein Structure Prediction competition), AlphaFold has become a major breakthrough in the field of protein structure prediction. AlphaFold 2 has achieved significant progress in predicting the structures of the human proteome, having predicted the three-dimensional structures of the vast majority of proteins in the human protein set. The impact of AlphaFold extends beyond basic research; it also offers new opportunities in drug discovery and design. AlphaFold's predicted structures can serve as starting points for drug design, helping to identify drug targets and compare with known ligand binding sites, thus accelerating the drug discovery process. Furthermore, the launch of AlphaFold 3 has expanded the frontiers of protein structure prediction to include the interactions of proteins with other biomolecules such as DNA, RNA, and small molecule ligands, potentially further advancing innovations in drug development.
Natural Language Processing (NLP) Model — ChemBERTa
ChemBERTa is a pre-trained natural language processing (NLP) model specially optimized for chemical text. It is based on the RoBERTa Transformer architecture and is pre-trained on chemical molecular data in Simplified Molecular Input Line Entry System (SMILES) format. ChemBERTa is capable of capturing the linguistic features of chemical molecules, thus playing a role in drug design. This project aims to advance drug discovery, chemical modeling, and property prediction, and has been showcased at multiple academic conferences, including Baylearn and the Royal Society of Chemistry's Chemical Science Symposium.
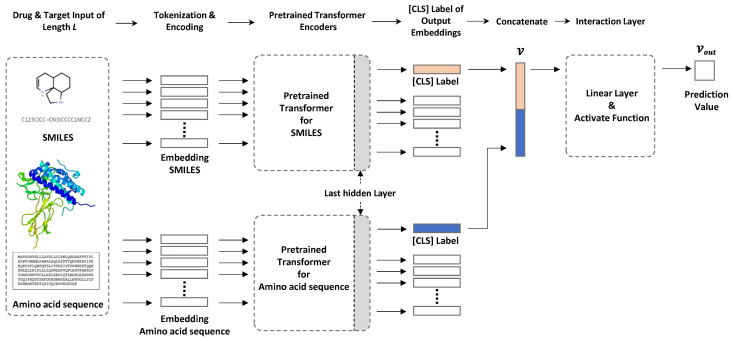
ChemBERTa undergoes pre-training on chemical datasets through a masked language modeling (MLM) task, such as on the ZINC 250k dataset, where the model is trained over 10 epochs until the loss converges to about 0.263. Additionally, ChemBERTa has been trained on subsets of PubChem of varying scales. Through HuggingFace, access is provided to various scales of pre-trained models, such as ZINC 100k, PubChem 100k, etc. The scalability of the pre-training datasets shows ChemBERTa's competitive downstream performance on MoleculeNet. ChemBERTa is widely used in the field of chemistry, including but not limited to drug discovery: using pre-trained models for drug property prediction, accelerating the drug R&D process; chemical synthesis route planning: leveraging the semantic understanding capabilities of Transformers to deduce the most effective synthesis routes; cheminformatics: for mining and classifying large-scale chemical databases, extracting key information; chemical research: assisting researchers in deciphering complex chemical reactions and compound structures. The introduction of pre-trained models has brought revolutionary changes to the field of chemistry. By pre-training on large-scale unlabeled corpora, ChemBERTa learns a general chemical representation, which is beneficial for downstream tasks. Pre-training also provides better model initialization, leading to better generalization performance and faster convergence speed on target tasks. Moreover, pre-training can serve as a regularization method to avoid overfitting on small datasets. The subsequent version, ChemBERTa-2, further expanded the scale of the pre-training dataset to 77 million compounds. ChemBERTa-2, by varying hyperparameters and pre-training dataset sizes, compared multi-task learning (MTR) and self-supervised pre-training (MLM), and pre-trained ChemBERTa-2 models on different pre-training tasks (MLM and MTR) and different dataset sizes (5M, 10M, and 77M) were compared against existing architectures on selected MoleculeNet tasks.
Hiro-LS: A Vertical Large Model in the Biopharmaceutical Field
Patsnap's large model application in the biopharmaceutical field, Hiro-LS, powered by PharmaGPT, provides a powerful data intelligence assistant for professionals in the biopharmaceutical field. PharmaGPT, with its 70 billion parameters, is capable of handling complex biopharmaceutical data and ensures professional accuracy in information retrieval. The model is developed using advanced natural language processing technology and extensive specialized data, covering key information across multiple stages from drug discovery to market analysis, enabling users to interact with the system through natural language and easily obtain the necessary patent literature, clinical trial data, industry policies, and other content.
The strength of Hiro-LS lies in its ability not only to perform intelligent retrieval but also to generate intuitive charts and tables based on users’ needs, making complex data straightforward at a glance. Users can quickly find relevant patent information by inputting chemical structures. Additionally, the system supports question-and-answer modes in both academic and patent domains, helping researchers to focus more on in-depth research work without having to waste time on tedious data organization.
Summary
From AlphaFold to Hiro-LS, these cutting-edge technological tools represent the latest achievements of artificial intelligence in the biopharmaceutical field. They not only greatly enhance the efficiency and success rate of drug discovery but also provide researchers with unprecedented insights, allowing them to examine the nature of diseases and their treatments from a broader perspective. Whether it’s AlphaFold's significant contribution to protein structure prediction or DeepPurpose's accurate modeling of drug-target interactions, both demonstrate the immense potential of deep learning technologies in future medical research.
The application of these tools also highlights the importance of interdisciplinary collaboration. The close cooperation between computer scientists and biologists has turned seemingly impossible tasks into achievable ones. As more similar platforms and technologies develop, we can look forward to witnessing further scientific discoveries and technological innovations emerging, collectively advancing human health.
For an experience with the large-scale biopharmaceutical model Hiro-LS, please click here for a quick and free trial of its features!
Reference
- 1. Huang, K. et al. DeepPurpose: a deep learning library for drug-target interaction prediction. Bioinformatics 36, 5545-5547 (2021). https://doi.org:10.1093/bioinformatics/btaa1005
- 2. Zhou, J. et al. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat Genet 50, 1171-1179 (2018). https://doi.org:10.1038/s41588-018-0160-6
- 3. Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493-500 (2024). https://doi.org:10.1038/s41586-024-07487-w
- 4. Kang, H. et al. Fine-tuning of BERT Model to Accurately Predict Drug-Target Interactions. Pharmaceutics 14 (2022). https://doi.org:10.3390/pharmaceutics14081710



