What is the Prospect of Lilly's BTK Inhibitor LOXO305?
In January, the FDA approved the first non-covalent BTK inhibitor, Pirtobrutinib (LOXO-305), for the market. This third-generation BTK inhibitor, developed by Eli Lilly, is approved for relapsed mantle cell lymphoma and refractory mantle cell lymphoma. Studies are ongoing for chronic lymphocytic leukemia (Phase III), non-Hodgkin lymphoma (Phase II), Waldenström's macroglobulinemia (Phase II), marginal zone lymphoma (Phase I/II), and hematologic malignancies (Phase I), etc.
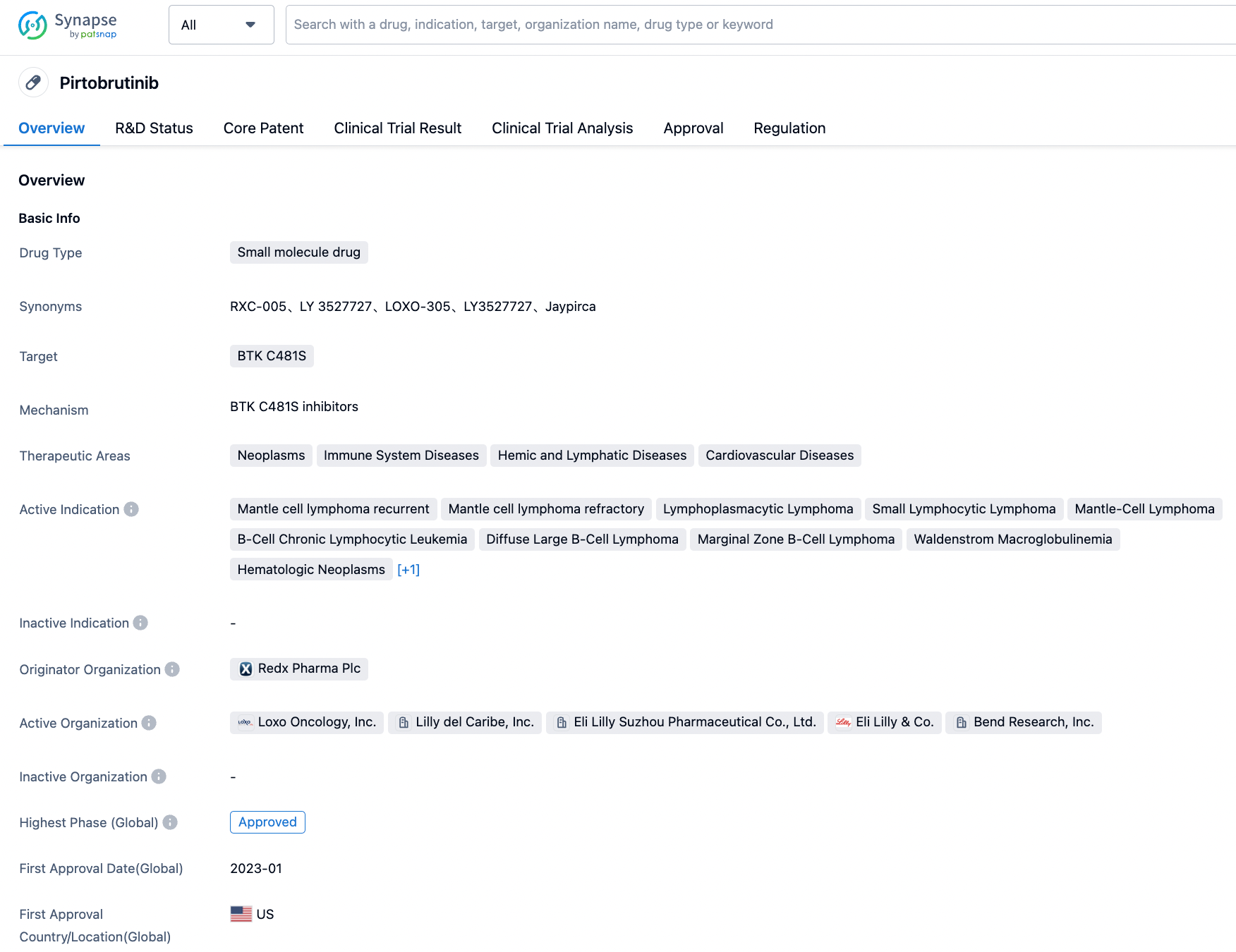
The Past and Present of Pirtobrutinib (LOXO-305)
Redx Pharma's BTK inhibitor project was sold to Loxo Oncology (later acquired by Eli Lilly) in July 2017 for $40 million. Pirtobrutinib (initially RXC005, then Loxo-305, now Jaypirca™) has been approved through the FDA’s accelerated approval pathway for the treatment of adult patients with relapsed or refractory mantle cell lymphoma (MCL) who have undergone at least two lines of systemic therapy, including a BTK inhibitor. RXC005 was successfully nominated as a candidate drug in October 2016, and preclinical data supporting this decision were presented at the 58th American Society of Hematology in December 2016 and at the 17th Chronic Lymphocytic Leukemia International Conference in May 2017.
FDA Accelerates Approval of Pirtobrutinib for MCL
Patients with lymphoproliferative disorders such as chronic lymphocytic leukemia and mantle cell lymphoma (MCL) who are resistant to covalent BTK inhibitors (cBTKis), especially those venetoclax-refractory, have unmet therapeutic needs. Pirtobrutinib can achieve a high response rate in patients with cBTKi refractoriness, regardless of the cBTKi resistance mechanism. This also led to the FDA accelerating approval for MCL in January. The toxicity profile of Pirtobrutinib suggests it is suitable for combination therapy, and combined clinical trials are ongoing.
The sales data has not yet been published, the future looks promising
According to the companies' financial reports, among the 6 inhibitors that have already been listed, apart from Ibrutinib, sales of other companies' products have achieved substantial growth in the first half of this year. In terms of sales and growth potential, the biggest threats to Ibrutinib are BeiGene's Zanubrutinib and AstraZeneca's Breztri. Eli Lilly's Q2 2023 financial report has not yet been released, however, in the Q1 2023 financial report, it was mentioned that new products contributed $573.6 million to sales in Q1 2023. These new products refer to those that have been launched since 2022, currently including pirtobrutinib and the GLP1 drug tirzepatide. Tirzepatide achieved sales revenues of $187 million in the third quarter of last year and considering the robust performance of GLP1 this year, the current half-year sales of pirtobrutinib do not exceed 20 million US dollars. Pirtobrutinib, after all, has just been launched and still has potential for the future.
BTK inhibitors (BTKis) are standard treatments for various B cell malignancies. The first-generation BTK inhibitor, ibrutinib (Imbruvica®), demonstrated the clinical importance of BTK as a drug target in many hematologic malignancies through its impressive clinical efficacy. The first three BTKis (ibrutinib, acalabrutinib, and zanubrutinib) bind covalently to the cysteine 481 (C481) residue of BTK, these are collectively referred to as covalent BTKis (cBTKis). All cBTKis are susceptible to resistance mutations in C481, the most common being C481S. For instance, ibrutinib has a short half-life, and the C481S mutation leads to insufficient BTK coverage within 24 hours, thereby affecting therapeutic efficacy. BTK C481S mutations are also common in patients with acalabrutinib and zanubrutinib-acquired resistance. Other mechanisms of resistance to cBTKis in chronic lymphocytic leukemia (CLL) include other BTK mutations, like gain-of-function mutations in the downstream kinase PLCG2. However, the mechanism of resistance is still unclear in up to 20% of CLL patients. In Waldenström's macroglobulinemia (WM), about 50% of patients develop BTK C481S mutations during disease progression, but these mutations are rare in mantle cell lymphoma (MCL) and marginal zone lymphoma, the resistance mechanism in these groups remains unclear.
Pirtobrutinib is a prominent member of the new generation of non-covalent BTKis, and its binding region is away from the C481 residue. Clinical trial data of pirtobrutinib, nemtabrutinib (MK1026, ARQ531), and vecabrutinib (SNS-062) showed significant clinical activity in patients whether with or without C481S mutations. The preclinical and early clinical trial results of pirtobrutinib support its recent accelerated approval.
The mechanism of action and pharmacology of Pirtobrutinib
X-ray crystal structures of BTK (wild-type and C481S) show that pirtobruinib coordinates on an adenosine triphosphate binding site distant from C481. Biochemical and cellular assays confirm that wild-type and C481S mutant BTK have the same binding and kinase inhibitory capabilities. As such, Pirtobrutinib can effectively inhibit autophosphorylation on Y223 of BTK. Surprisingly, as a back-pocket binder, pirtobrutinib also inhibits phosphorylation at the Y551 site of upstream BTK,fwhich may be due to pirtobrutinib stabilizing BTK in a closed, inactive conformation. This structural consideration is significant, as Y551 activation is crucial for "kinase-dead" BTK mutants to recruit hematopoietic cell kinase (HCK), which could explain how these mutants can still signal even when the kinase site is deactivated.
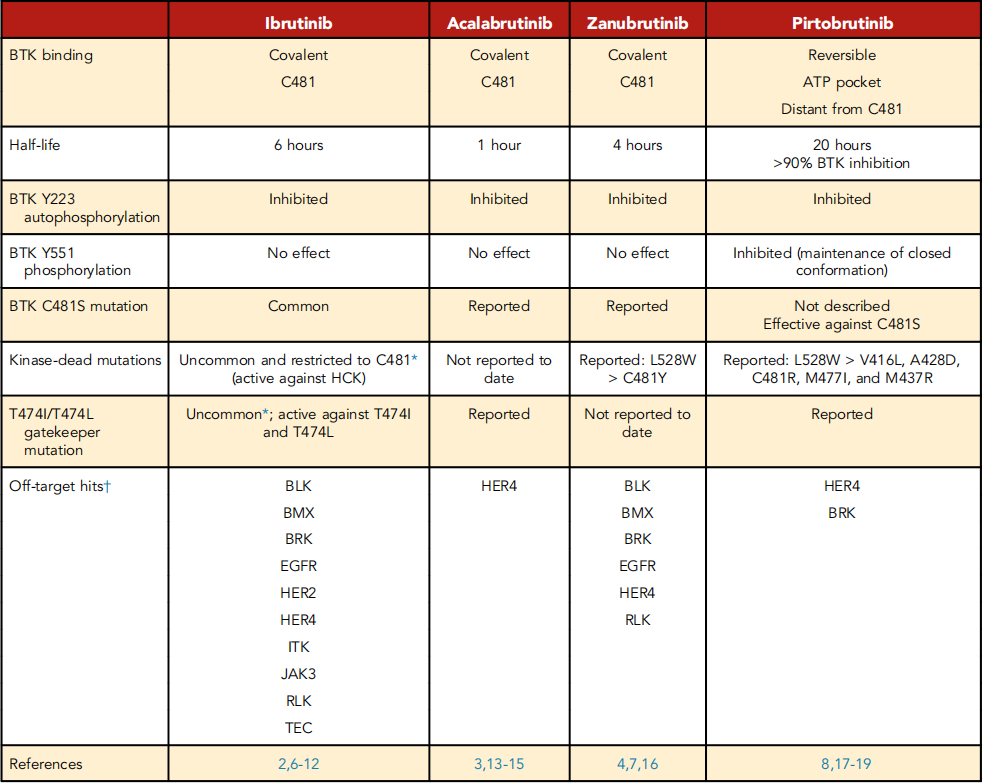
Biochemical tests and cellular assays confirm the high selectiveness of pirtobrutinib on BTK, with the number of off-target hits far less than Ibrutinib and Zanubrutinib. The only kinases affected by pirtobrutinib by less than 20 time folds are HER4 and BRK. Research shows that the selectivity of cBTKis is related to reducing cardiovascular and other adverse events (AE).
In the first in-human study, pirtobrutinib demonstrated linear dose-proportional exposure in the test dosage range of 25mg to 300mg daily and has a half-life of 20 hours. No dose-limiting toxicity was observed and the recommended stage 2 dosage (200mg daily) was selected based on the trough plasma concentration that can achieve 96% BTK inhibition. Therefore, the long half-life of pirtobrutinib provides continuous BTK target occupancy, which is different from cBTKis, where irreversible binding compensate for the short half-life (Table 1). A theoretical advantage of a long half-life is that newly synthesized BTK molecules within a 24-hour window are also subject to inhibition.
Clinical Data of Pirtobruinib
BRUIN Trial
Phase 1/2 BRUIN trial involved 773 patients with relapsed/refractory (R/R) B-cell malignancies. Most adverse events (AEs) were low-grade. Neutropenia (20.4%) was the most common grade ≥3 AE occurring post-treatment. Only 2.6% patients discontinued treatment due to adverse reactions. As for special AEs, the occurrence rate of grade ≥3 bleeding was 1.8%, any grade hypertension was 2.3%, and atrial fibrillation/flutter was 2.8%.
The expansion cohort included patients with chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), mantle cell lymphoma (MCL), Waldenstrom’s macroglobulinemia (WM) and marginal zone lymphoma previously treated with irreversible BTK inhibitor (cBTKi), as well as Richter Transformation (RT) patients.
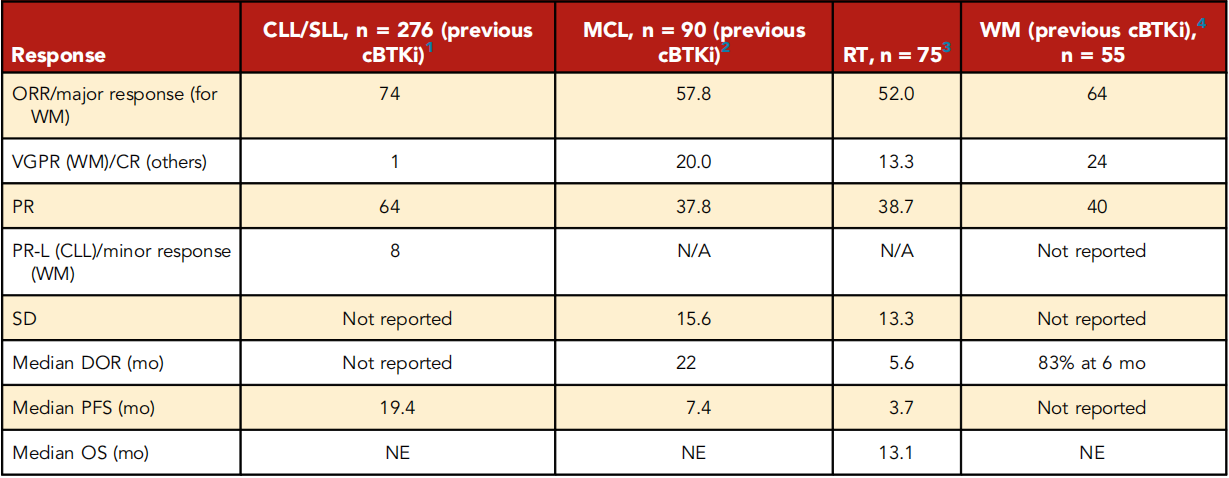
MCL: The survival period of MCL patients who progress or are intolerant during cBTKi treatment before chimeric antigen receptor T-cell therapy is 3 to 10 months. Brexucabtagene ciloleucel is a drug approved by the FDA for this population based on the ZUMA2 study, with durable remissions in 40% to 50% of patients, but it is toxic and impractical for many patients. Additionally, real-world data indicates that median progression-free survival (PFS) in TP53 mutant patients is less than 6 months. The latest update of BRUIN MCL data focused on 90 patients previously exposed to cBTKis, with a median of 3 prior treatments, and 82% of patients discontinued cBTKi due to disease progression. The final data showed an overall response rate (ORR) of 58% (complete remission, 20%) after pirtobrutinib treatment; the median duration of response (DOR) in 52 responsive patients was 22 months. In patients who discontinued BTKi due to disease progression, ORR and DOR were lower (ORR, 50%; median DOR, 14.8 months). These data prompted the FDA to accelerate the approval of pirtobrutinib for MCL patients previously treated at least twice, including with cBTKi.
CLL/SLL: There are no recognized standard treatment methods for CLL/SLL patients (double drug-resistant) resistant to both cBTKi and venetoclax, especially for pentad drug-resistant patients (prior cBTKi, chemotherapy, CD20 antibody, BCL2i and PI3Ki). Among the 247 cBTKi-treated patients announced at the 2022 American Society of Hematology Annual Meeting, pirtobrutinib had an ORR of 82% among CLL patients; all patients had previously received cBTKi treatment. The ORR for double drug-resistant patients (previously treated 5 times) was 79% (median PFS, 16.8 months), and for the pentad drug-resistant patients was 77.8%. The entire cohort had a median PFS of 19.4 months (16.8 months for double drug-resistant patients). Preclinical data predicts Pirtobrutinib to be effective in patients with BTK C481S mutants; in addition, ORR was 73.9% in patients without BTK C481 mutation, and 55.6% in patients with activated PLCG2 downstream mutation, which reduces the disease's dependence on BTK.
Chemotherapy: Chemotherapy immunotherapy is ineffective and toxic for most cancer patients. Among the 75 evaluable patients (median of 4 times for CLL and RT treatment), Pirtobrutinib had an ORR of 52%, including 13.3% of complete remissions. The median DOR was 5.6 months. 6 patients underwent allogeneic stem cell transplantation. It is recommended for evaluation in larger cohorts and combined approaches.
WM: A total of 78 WM patients were treated, with 78% of patients previously receiving cBTKis, and 64% receiving chemotherapy, CD20 antibodies, and cBTKis. Among patients previously treated with cBTKi (66% discontinued due to progression), ORR was 64%, and 6-month PFS for responders was 83%.
In the phase 1/2 BRUIN trial of 773 patients, 127 were intolerant to previous cBTKi (95% to ibrutinib), the most common reason being atrial fibrillation/flutter (24%). Pirtobrutinib was well tolerated, with 7 out of 127 discontinuing due to Pirtobrutinib-related AE, and no discontinuations due to recurrence of previous cBTKi-related AE. In this subgroup, ORR was 77% for CLL/SLL and 82% for MCL, with a median DOR of 28.4 months for CLL/SLL and MCL was not estimable.
The Drug Resistance Mechanism of Pirtobrutinib
Reports indicate that drug resistance persists, with two recent reports emphasising the btk mutations (other than c481s) in advanced patients treated with Pirtobrutinib. Surprisingly, these BTK mutants are kinase-dead mutants, possessing inactive kinase sites that are unable to self-phosphorylate Y223, but still exhibit active downstream signaling. These kinase-dead mutants are rarely seen in ibrutinib-resistant patients but are relatively common in patients treated with zanubrutinib or Pirtobrutinib. These differences may be due to the off-target effect of Ibrutinib on HCK3; the recruitment of HCK may explain how these kinase-dead BTK mutants continue to signal. Reports also exist of the T474 Gatekeeper mutation in patients with progressive disease after treatment with acalabrutinib and Pirtobrutinib.
Future Directions
Pirtobrutinib is the first commercially available non-covalent BTK inhibitor approved by the FDA for use in MCL patients who have previously received cBTKi treatment. Expanded access schemes specific to each country are also available. Evaluations of the application of Pirtobrutinib in early treatment and head-to-head Phase 3 studies with cBTK inhibitors are currently underway.
As shown in the following figure, in the clinical trials and the primary endpoints, a Phase 3 trial in R/R MCL compared Pirtobrutinib with several cBTKi's chosen by investigators. The Phase 3 development program for CLL includes four studies: two studies evaluate Pirtobrutinib in comparison to 1L (Bendamustine and Rituximab (BR)) and R/R (BR or Idelalisib + Rituximab) standard treatment regimens; one compares Pirtobrutinib with Ibrutinib in terms of efficacy in 1L and R/R CLL; the fourth compares triple therapy of Pirtobrutinib + Venetoclax + Rituximab with Venetoclax + Rituximab (VR) in cases of R/R CLL.
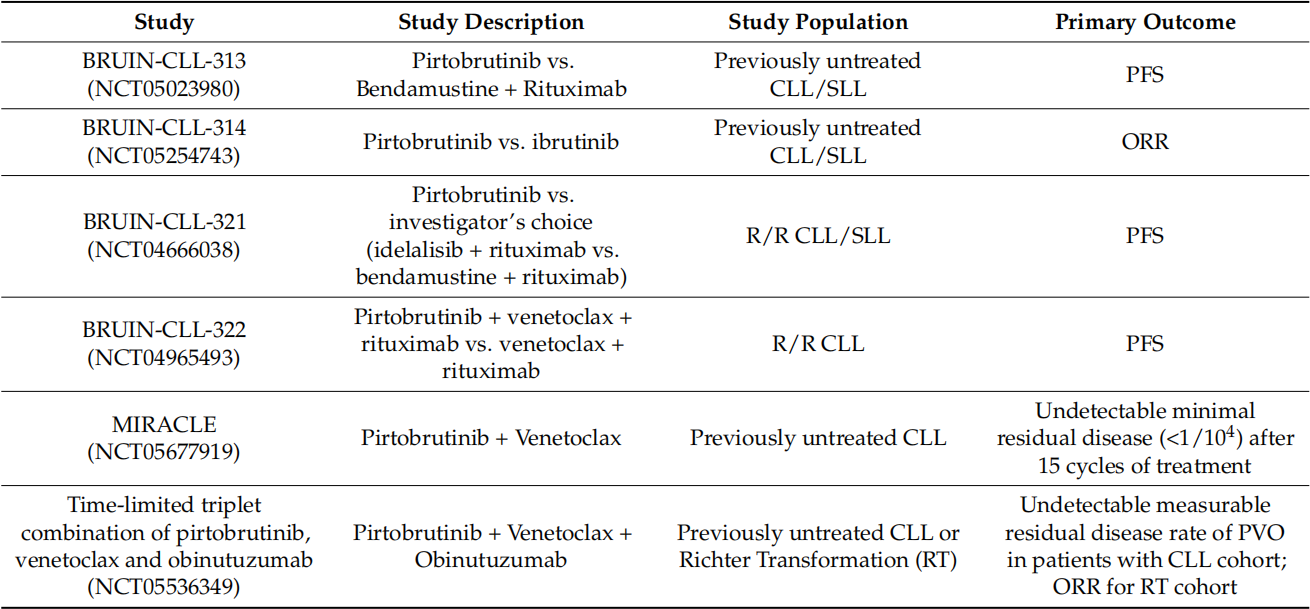
We are looking forward to the disclosure of subsequent clinical results of pirtobrutinib, and the data from several head-to-head Phase 3 clinical trials will also have a significant impact on the fate of pirtobrutinib. There have been quite a few cases where early approval was granted through accelerated approval, but the results of subsequent clinical trials were not satisfactory. Therefore, while we look forward to this, we should also reserve some caution.
While paying attention to the third-generation non-covalent BTK inhibitors, we can also consider other approaches to solve drug resistance. This requires a deeper understanding of the mechanisms of acquired resistance to cBTKi and ncBTKi, as well as the possibility of cross-resistance between cBTKi and ncBTKi. In addition, preclinical and clinical studies of new combination therapies and new treatment strategies (such as BTK degraders, bispecific antibodies, and pkc-β inhibitors) may also be crucial to improving patients' prognosis in the future.

Reference
1.Constantine S. Tam et.al; Pirtobrutinib: a new hope for patients with BTK inhibitor–refractory lymphoproliferative disorders. DOI: 10.1182/blood.2023020240.
2.Skye Montoya et al;Non-Covalent Bruton’s Tyrosine Kinase Inhibitors in the Treatment of Chronic Lymphocytic Leukemia. Cancers 2023, 15, 3648.
3.Wang ML, et al. Efficacy of pirtobrutinib in covalent BTK-inhibitor pre-treated relapsed / refractory mantle cell lymphoma: additional patients and extended follow-up from the phase 1/2 BRUIN study [abstract]. Blood. 2022;140(suppl 1):9368-9372.
4.US Food and Drug Administration. FDA grants accelerated approval to pirtobrutinib for relapsed or refractory mantle cell lymphoma; 2023. Accessed 15 February 2023.
5.https://mp.weixin.qq.com/s/ZJ5VFpH7PG5FdJV9m0mk5g.



