Wang Shaomeng's research group reports AR receptor PROTAC drug ARD-1676, with a bioavailability as high as 99%
Due to their large molecular weight (700-1200Da), poor solubility, and low permeability, PROTAC molecules generally have a low oral bioavailability (F). Therefore, researchers often go to great lengths to increase F, such as optimizing the linker to improve permeability and metabolic stability, selecting E3 ligase ligands with smaller molecular weights, and using prodrug strategies, etc.
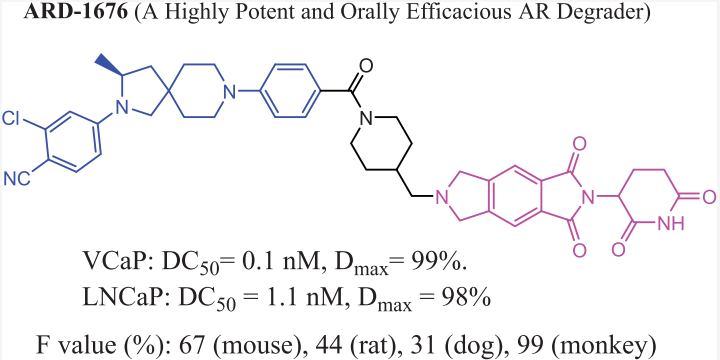
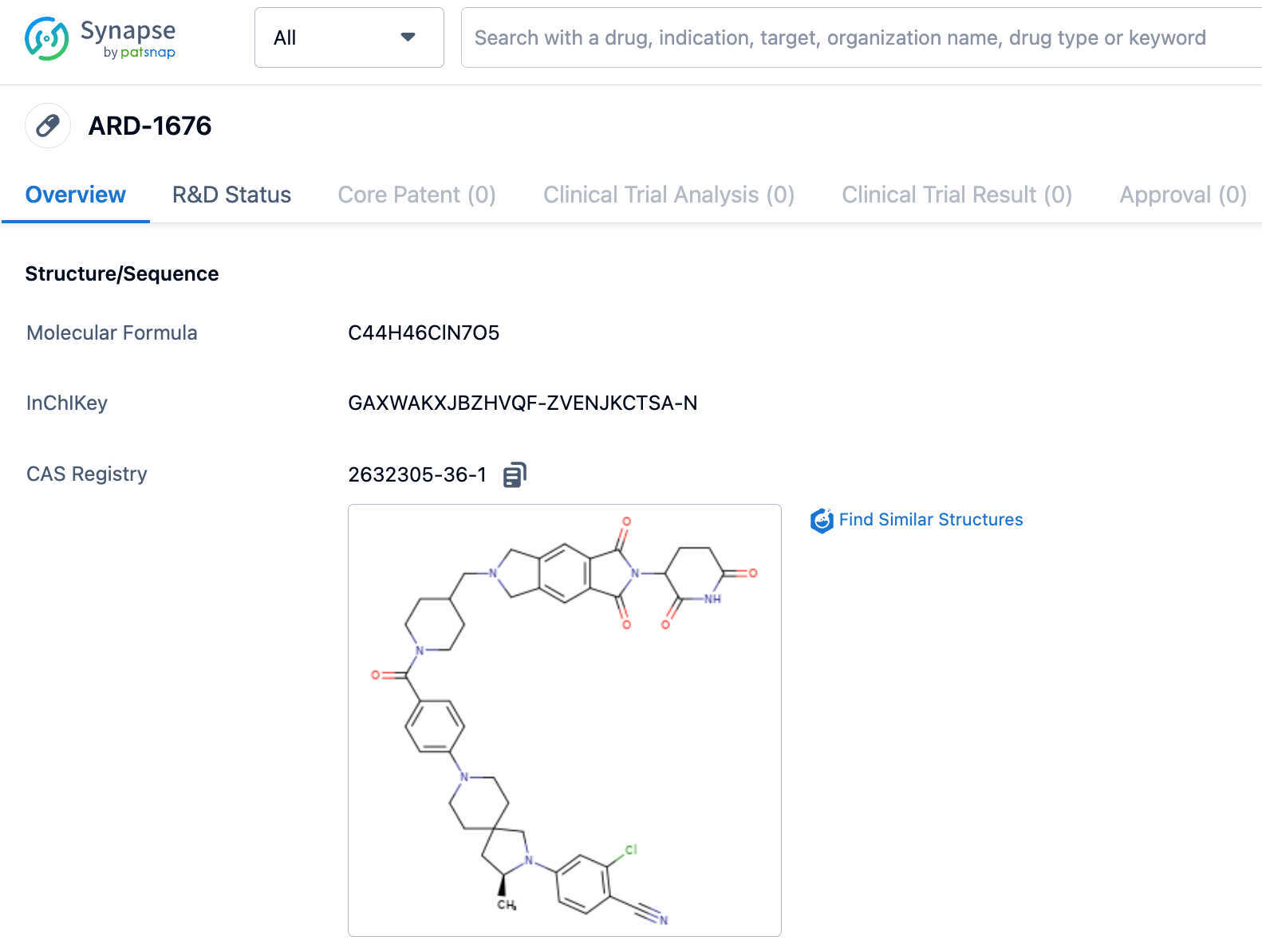
The research group of Wang Shaomeng recently published a paper in JMC, reporting the discovery of ARD-1676 and characterizing its preclinical properties. ARD-1676 is an orally effective PROTAC androgen receptor (AR) degrader. The paper mentions that the DC50 values of ARD-1676 in AR+ VCaP and LNCaP cell lines are 0.1 and 1.1 nM respectively, and the IC50 values in VCaP and LNCaP cell lines are 11.5 and 2.8 nM respectively.
👇Please click on the image below to directly access the latest data (R&D Status | Core Patent | Clinical Trial | Approval status in Global countries) of this drug.
ARD-1676 can effectively induce the degradation of a series of clinically relevant AR mutants. The oral bioavailability of ARD-1676 in mice, rats, dogs, and monkeys is 67%, 44%, 31%, and 99% respectively. Oral administration of ARD-1676 can effectively reduce the AR protein levels in mouse VCaP tumor tissues and inhibit tumor growth in a VCaP mouse xenograft tumor model, with no signs of toxicity.
The oral bioavailability in monkeys is 99%, which is relatively rare among PROTAC molecules.
The ADME Characteristics of ARD-1676
ARD-1676 has lower passive cell permeability, with Papp A-B not reaching 1×10-6, and an efflux ratio of 1.8, suggesting a low potential as a substrate for P-glycoprotein (Pgp). The study evaluated the stability of ARD-1676 in the liver microsomes, hepatocytes, and plasma in mice, rats, dogs, non-human primates (NHP), and humans. It was concluded that the compound demonstrated good stability in the microsome, hepatocytes, and plasma from all five species.
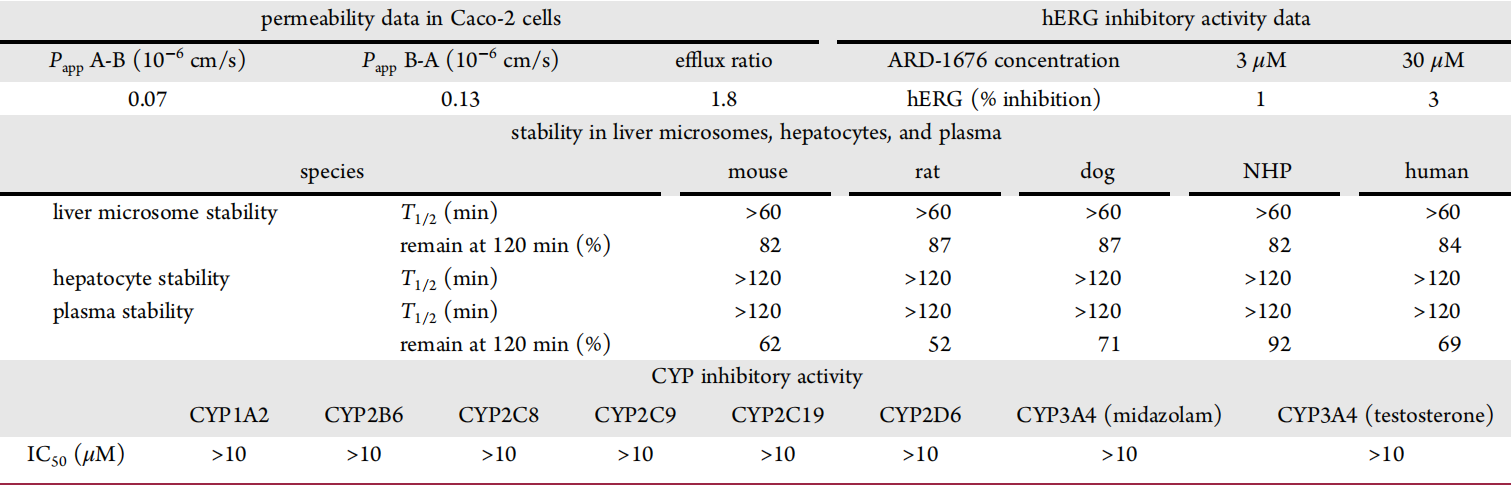
The study also evaluated ARD-1676 for its inhibitory effect on all major cytochrome P450 (CYP) subtypes and found that ARD-1676 had no inhibitory effect on the evaluated CYP subtypes. The potential inhibitory effect of ARD-1676 on hERG in vitro was also assessed. Compared to the control group, the inhibitory effect of ARD-1676 under 3 and 30 μM was negligible, suggesting that ARD-1676 does not have a hERG toxicity trend.
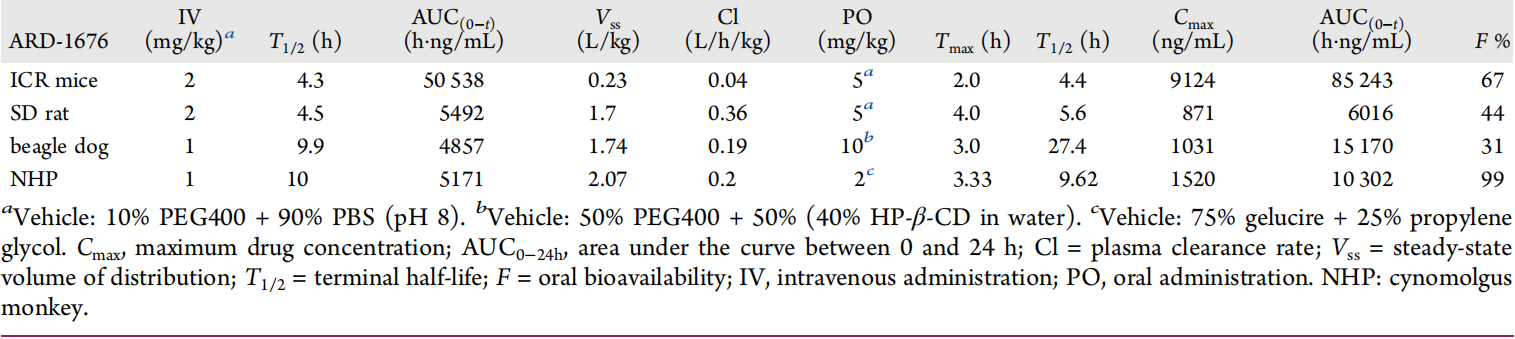
The figure above shows the pharmacokinetic parameters of ARD-1676 in mice, rats, dogs, and monkeys. In mice, ARD-1676 has a very low clearance rate (Cl = 0.04 L/h/kg), a moderate volume of distribution (Vss = 0.23 L/kg), and a T1/2 of 4.3-4.4 h for both intravenous and oral administration. With a dose of 5 mg/kg, the Cmax = 9124 ng/mL, AUC = 85243 h·ng/mL, and the oral bioavailability is 67%.
In dogs, ARD-1676 has a low clearance rate (Cl = 0.19 L/h/kg), a moderate volume of distribution (Vss = 1.7 L/kg), an intravenous T1/2 of 9.9 h, an oral T1/2 of 27.4 h, a Cmax of 1031 ng/mL for oral administration, and an AUC = 15170 h·ng/mL. The oral bioavailability is 31%.
In monkeys, ARD-1676 has a low clearance (Cl = 0.2 L/h/kg), a moderate volume of distribution (Vss = 2.1 L/kg), and a T1/2 of 9.6 ~ 9.9 h for both intravenous and oral administration. With a dose of 2mg /kg, the Cmax = 1520 ng/mL, AUC = 10302 h·ng/mL, and the oral bioavailability is 99%.
It is worth noting that the solvents used across the four species are different, which can greatly impact bioavailability.
The Pharmacokinetics and Antitumor Effect of ARD-1676 in Mice
A single oral dose of 12.5 mg/kg of ARD-1676 was administered to mice with VCaP xenograft tumors. The immunoblot analysis of tumor tissues showed that ARD-1676 effectively decreased the level of AR proteins by 96% and 93% at the 6 h and 24 h time points, respectively. According to the pharmacodynamic data of ARD-1676, its antitumor activity was determined in the VCaP xenograft tumor model in mice, yielding the data graphically represented below. The pharmacokinetics data indicated that ARD-1676 effectively and dose-dependently inhibited tumor growth under all 3 assessed doses.
ARD-2051 and ARD-1676
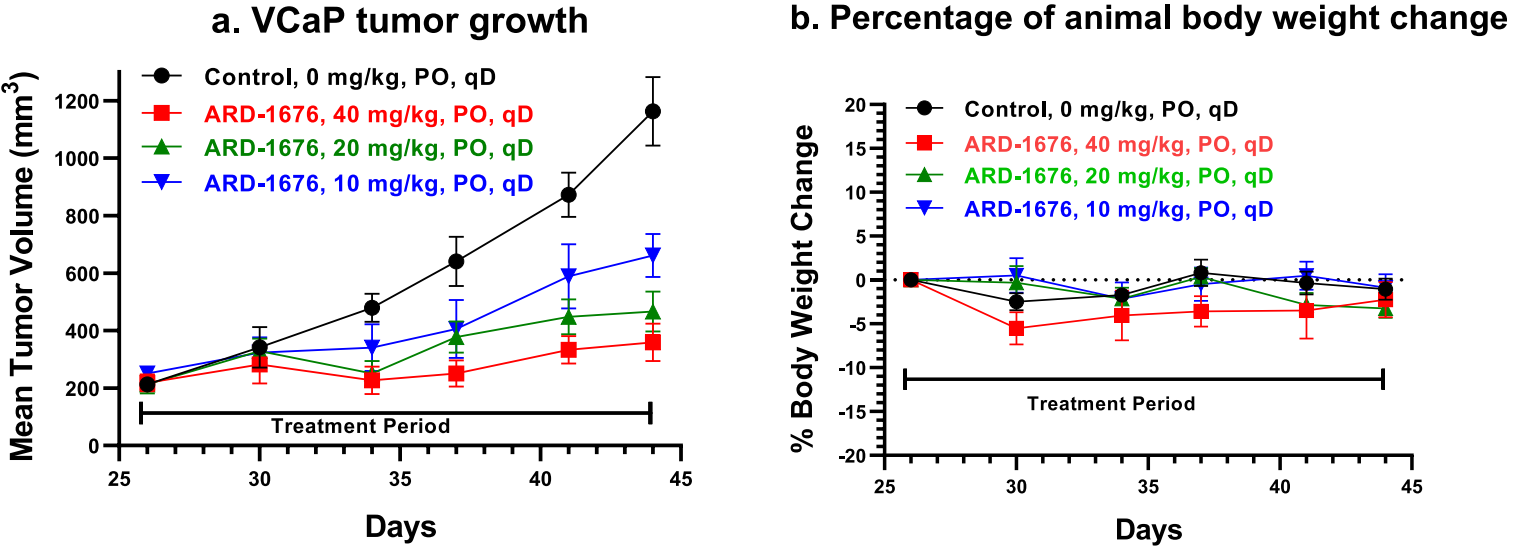
In June this year, the team led by Wang Shaomeng also reported the discovery of ARD-2051. As shown in the figure below, the oral bioavailability of ARD-2051 is relatively high in small animals, but not enough in large animals. From the comparison of 27 and 30, it can be seen that the chiral methyl on the cyclobutene ring has a very significant impact on the degradation efficiency. Furthermore, the synthesis of this chiral methyl is challenging, which requires the opening and reclosing of substituted epoxide into 5-membered ring. The linker was further optimized to obtain ARD-1676.
Progress in the Development of AR Targeting PROTAC Drugs
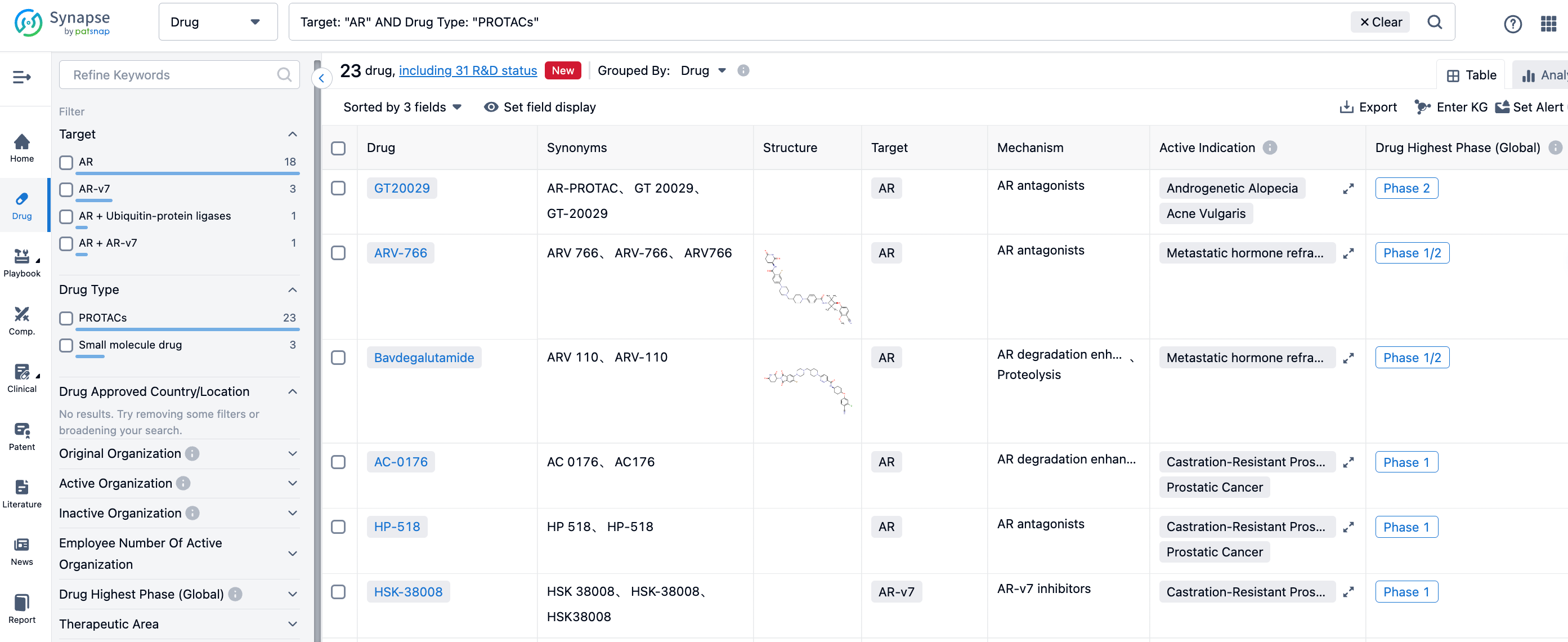
In the Synapse database, 23 drugs were found when searching for AR and PROTAC, involving 31 development statuses, as shown in the above figure, numerous pharmaceutical companies are participating in this heated competition. Among them, GT20029 from Suzhou Kintor Pharmaceuticals is at Phase II of clinical trials, is being studied for indications such as androgenic alopecia and acne vulgaris; Arvinas' ARV-766 and ARV-110 are both in Phase I/II, with indications under research being metastatic hormone-refractory prostate cancer; Accutar Biotechnology's AC-0176, Hinova Pharmaceuticals' HP-518, and Haisco Pharmaceutical's HSK-38008 are all undergoing Phase I clinical trials, and the indications under study are castration-resistant prostate cancer and prostate cancer.
The bioavailability of PROTACs
The bioavailability of PROTACs is generally poor. Lipinski's Rule of Five (Ro5) is an empirical rule that sets several criteria for drug bioavailability, typically used to predict the bioavailability of compounds. With the advent of small molecule PROTACs, the molecular weight of PROTACs has been reduced, but the hetero-bifunctional structure of PROTACs results in their violation of Ro5, leading to poor bioavailability. Permeability tests have shown that most PROTAC molecules have poor cellular permeability, thus, to date, most in vivo studies have adopted parenteral administration.
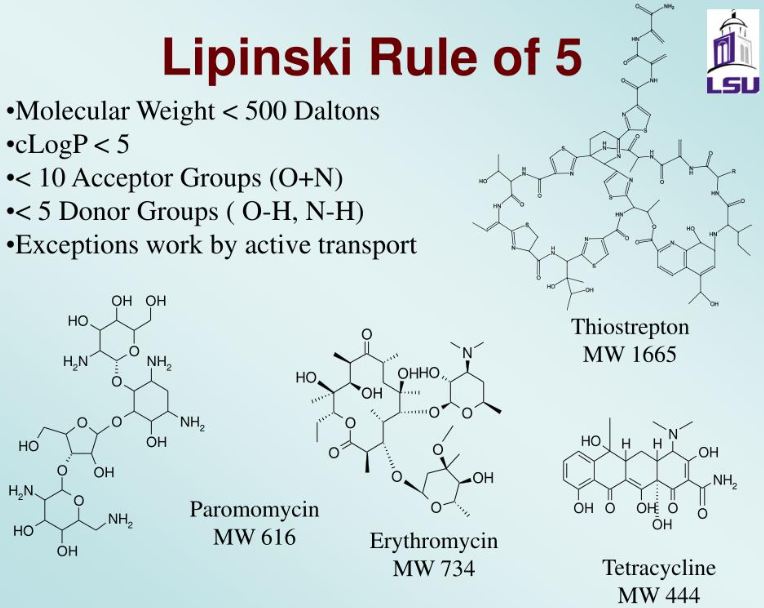
As of 2017, approximately 6% of orally administered drugs were not within the Rule of 5 (Ro5) scope. For example, many macrocycles can be given orally, even though they clearly exist within the “beyond Rule of 5 (bRo5)” chemical space. This could be explained by their chameleon-like conformational flexibility, which promotes intestinal absorption of drugs.
Pharmaceutical companies and research institutions are developing oral PROTACs, urgently requiring reasonable design standards to endow PROTACs with oral effectiveness. A study on the relationship between physicochemical properties and permeability conducted by Lokey and his colleagues has found that, even in PROTACs with almost the same molecular weight, the same ALogP, and the same number of hydrogen bond donors/acceptors, cell permeability could differ by a factor of 10. Moreover, reducing the number of hydrogen bond donors/acceptors usually improves the bioavailability of small molecules, but in the case of PROTACs, it may result in worse solubility. One milestone study by the Lokey group has showed the effectiveness of using intramolecular hydrogen bonds to mask hydrogen bond donors/acceptors, based on the aforementioned conformational flexibility. However, the rational design of such compounds with intramolecular hydrogen bonds is highly challenging.
Summary
Androgen receptors (AR) and AR signaling play a crucial role in the initiation and progression of human prostate cancer. AR-targeted therapeutic drugs, including abiraterone which inhibits androgen synthesis, and second-generation AR antagonists like enzalutamide, apalutamide and darolutamide, have been developed for the treatment of advanced human prostate cancer. Even though these drugs have proved to be effective in clinical practice, resistance to them develops within 18 months. Key resistance mechanisms include AR gene amplification, activating AR mutations, and expressions of AR variants. In most human prostate cancers resistant to AR-targeted drugs, AR and AR signaling continue to feature in tumor progression, prompting the search for new therapeutic strategies targeting AR and AR signaling.
An appealing new strategy is to target AR through induced degradation of target proteins. Inspired by the clinical success of selective estrogen receptor degraders (SERDs) in the treatment of human breast cancer, selective AR degraders (SARDs) are under investigation. However, the degradation capacity of current SARD molecules is relatively weak and needs significant improvement.
A second method to induce AR protein degradation is the use of PROTAC technology. PROTAC drugs include a ligand that binds to the target protein, a ligand that binds to and recruits the E3 ligase or E3 ligase complex, and a linker that connects them. The first AR PROTAC molecule was designed using bicalutamide derivatives as the AR ligand and MDM2 inhibitors as the E3 ligase ligand.
With companies like Arvinas and various academic institutions continually advancing PROTAC technology, it is hoped that this kind of drug will be introduced to the market as early as possible to overcome drug resistance, thus enriching options for patients' medication.

References
1.Shaomeng Wang et.al; Discovery of ARD-1676 as a Highly Potent and Orally Efficacious AR PROTAC Degrader with a Broad Activity against AR Mutants for the Treatment of AR + Human Prostate Cancer.https://doi.org/10.1021/acs.jmedchem.3c01264.
2.Shaomeng Wang et.al; Discovery of ARD-2051 as a Potent and Orally Efficacious Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor for the Treatment of Advanced Prostate Cancer.J. Med. Chem. 2023, 66, 8822−8843.
3.Minoru Ishikawa et.al; In vivo synthetic chemistry of proteolysis targeting chimeras (PROTACs).Bioorg. Med. Chem. 41 (2021) 116221.



